So very few of us working in ICUs did not walk around with this book in our lab coat pocket – no wait, that was the 90’s so in the last decade it would have been as a pdf on a phone…
So it’s been a true pleasure to meet him and exchange, but we are certainly looking forward to him joining the H&R family, which has been such a nidus of individual growth and amazing collaborations over the years. Please note that Dr. Marino is still a practicing intensivist and so an example to us all!
So it’s always interesting to unpack a new set of guidelines, and while we were not overly optimistic, we were still a bit surprised about the (lack of) evolution for the 2026 edition of Surviving Sepsis. Firstly would like to acknowledge the tremendous amount of work and organization by the SSG which have greatly contributed to awareness of sepsis and in that way certainly saved many lives.
In this set, the first thing we glitched on was a particulat statement about MAP, which we felt was, physiologically, simply incorrect. So we had a chat about it today.
We have a lot more to unpack but we’re putting that in an article that we’ll be submitting by next week.
Of course, if you like our bedside physiology way of approaching patients as we eschew one-size-fits-all, there is no better place to learn bedside diagnostic skills and physiological reasoning as HR. Join us in Montreal next september!
Pour la prochaine édition de notre conférence sesquiennale, Foundations Reimagined: Physiology at the Bedside, nous proposons une immersion approfondie dans des principes physiologiques fondamentaux souvent mal compris — mais pourtant essentiels au chevet du patient. Grâce à des conférences ancrées dans la pratique clinique et à un apprentissage interactif basé sur des cas, les participants repartiront avec un cadre durable pour aborder le patient en phase aiguë.
Nous nous éloignons progressivement d’une ère de médecine rigide fondée sur des protocoles pour entrer dans celle de la précision et des soins personnalisés. Bien que cette évolution promette de meilleurs résultats pour les patients, elle exige également davantage du clinicien au chevet : une compréhension plus approfondie, des compétences plus aiguisées et une plus grande confiance — plutôt que de simplement appliquer un protocole de manière automatique ou extrapoler des données populationnelles au patient unique devant nous.
À la suite de notre événement précédent introduisant le concept des interfaces hémodynamiques, il est apparu clairement que les cliniciens ont besoin d’une base physiologique plus solide, combinée à des discussions de cas interactives, afin de développer une approche pratique et utilisable de l’évaluation et de la prise en charge du choc. Étant donné le chevauchement important entre le choc et l’insuffisance respiratoire, cette conférence proposera également une approche physiologique complète de l’insuffisance respiratoire.
Des sessions avancées d’hémodynamique en POCUS seront offertes à ceux qui souhaitent évoluer à la fine pointe, parallèlement à des sessions fondamentales de POCUS conçues pour soutenir les apprenants plus tôt dans leur parcours.
En septembre 2026, nous réunirons de nouveau un corps professoral international composé de penseurs et d’éducateurs d’élite afin d’offrir une physiologie appliquée, des compétences au chevet du patient et un raisonnement clinique transposables en salle d’hospitalisation, au bloc opératoire, à l’urgence et à l’unité de soins intensifs.
Incorporant comme toujours un grouoe grandissant d’educateurs internationaux hors-pair et incluant des quasi-legendes de soins intensifs tels Dr. Andre Denault et Dr. Sheldon Magder, nous aurons cette anne le plaisir d’avoir en presentiel non seulement le Dr. Scott Weingart mais aussi pour la premiere fois Paul Marino, du fameux livre “The ICU Book,” un livre virtuellement symbolique des soins intensifs! Ne manquez pas la chance de venir apprendre et echanger avec eux!
Ne manquez pas cette occasion exceptionnelle d’apprendre, de partager, de bâtir et de faire partie d’un groupe remarquable uni par un objectif commun : faire progresser la médecine.
Comite Scientifique
Gigi Liu · Andre Denault · Ben Daxon · Ian Ajmo · Kiran Rikhraj · Jasmine Lam · Julien Viau-Lapointe · Philippe Rola
Preliminary Programme
DAY 1
Single Track – Morning
Time
Session
07:55–08:00
Welcome – Rola & Spiegel
08:00–08:30
Introduction & Clinical Case – Crager
08:30–09:00
How the Blood Goes ‘Round – Miller
09:00–09:30
Andromeda 2: What Did We Learn? – Hernandez
09:30–10:00
Understanding RCT Limitations at the Bedside – Lynn
10:00–10:30
Coffee Break
10:30–11:00
Hot topic TBA!
11:00–11:30
RV at the Bedside – Siuba
11:30–12:00
“Mind of the Expert” Case Discussions – Panel
12:00–13:00
Lunch
13:00–13:45
Keynote Lecture: Changing the Oxygen Paradigm – Marino
Split Tracks – Afternoon
Respiratory Track
Time
Session
13:45–14:15
Understanding FRC – Daxon
14:15–14:45
Managing the First Hour of Severe Respiratory Failure –
14:45–15:15
Managing the Next 24 Hours of Severe Respiratory Failure – Spiegel
15:15–15:30
Coffee Break
15:30–17:00
Workshops
Extreme Hemodynamics Track
Time
Session
13:45–14:00
The RV Waveform: Clinical Applications – Denault
14:00–14:15
Understanding Afterload – Magder
14:15–14:30
Renal Hemodynamic Manipulation – Corradi
14:30–15:00
Surfing the Spectrum of Tamponade – Scott
15:00–15:30
Coffee Break
15:30–17:00
Workshops
DAY 2
Single Track – Morning (Case-Based with Workshops)
Time
Session
08:00–08:20
Case 1 – Miller & Haycock
08:20–08:40
Case 2 – Spiegel & Hockstein
08:40–09:00
Case 3 – Denault & Scott
09:00–09:20
Case 4 – Crager & TBA
09:20–10:40
Workshops & Coffee (VTI / EF / TAPSE / VExUS)
10:40–11:00
Case 5 – Kattan & TBA
11:00–11:20
Case 6 – Argaiz & TBA
11:20–11:40
Case 7 – Siuba & TBA
11:40–12:00
Case 8
12:00–12:30
Keynote: PreOx 2026 – Weingart
12:30–13:30
Lunch
Split Tracks – Afternoon
Respiratory Track
Time
Session
13:30–14:00
Navigating NIV Choices – TBA
14:00–14:20
Can I Give Inhaled Drugs to NIV Patients?
14:20–14:40
Approaching Vent Dyssynchrony – Chatterjee or Mireles-Cabodevila
14:40–15:40
When Do I Reach for Inhaled Drugs in Intubated Patients? – Daxon
15:40–16:30
Workshops & Coffee
16:30–17:00
CPAP as a Mode of Ventilation – Spiegel
17:00–17:30
Thoracic Electrical Impedance in ALI– Viau-Lapointe
Extreme Hemodynamics Track
Time
Session
13:30–14:00
The Doppler Envelope: Fundamentals – Argaiz
14:00–14:20
Resuscitating Aortic Stenosis – Augustin
14:20–14:40
The Modified Tei Index – Haycock
14:40–16:00
Workshops & Coffee
16:00–16:30
Using Cerebral Oximetry to Tailor Resuscitation – Denault
Posting this for my good friend Jeff Scott who runs an awesome unit in sunny Miami! Jeff has long been part of the HR family and I wish I could work in his shop myself!
“CVICU-SHOCK” & Lung Rescue – Transplant Units
Familiar with Ecmo, LVADs, Impella and this specific patient population.
In the next edition of our sesquiennial conference, Foundations Reimagined: Physiology at the Bedside, we propose a deep dive into core physiological principles that are often misunderstood—but critically important at the bedside. Through clinically grounded lectures and interactive, case-based learning, participants will leave with a durable framework for approaching the acutely ill patient.
We are steadily moving away from an era of rigid, protocol-based medicine toward one of precision and personalized care. While this evolution promises better patient outcomes, it also demands more from the bedside clinician: deeper understanding, sharper skills, and greater confidence—rather than simply rubber-stamping a protocol or extrapolating population-level data to the single patient in front of us.
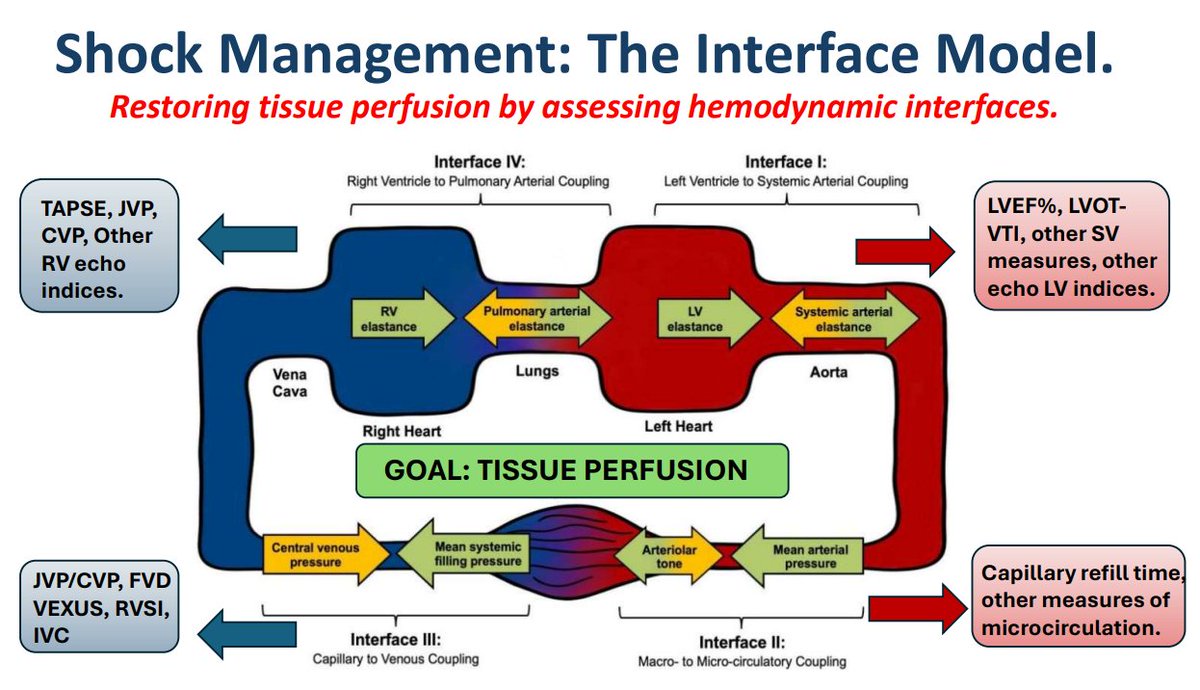
Following our previous event introducing the concept of hemodynamic interfaces, it became clear that clinicians need a stronger physiological foundation combined with interactive case discussions to develop a practical, usable approach to shock assessment and management. Given the profound overlap between shock and respiratory failure, this conference will also feature a comprehensive physiologic approach to respiratory failure.
Advanced POCUS hemodynamics sessions will be offered for those seeking to operate at the cutting edge, alongside parallel core POCUS sessions designed to support learners earlier in their journey.
In September 2026, we will once again assemble an international faculty of elite thinkers and educators to deliver practical physiology, bedside skills, and clinical reasoning applicable across the wards, operating room, emergency department, and ICU.
Don’t miss this amazing opportunity to learn, share, build and belong to an outstanding group of people who share the common goal of moving the needle in medicine.
Scientific Committee
Gigi Liu · Andre Denault · Ben Daxon · Ian Ajmo · Kiran Rikhraj · Jasmine Lam · Julien Viau-Lapointe · Philippe Rola
Preliminary Programme
DAY 1
Single Track – Morning
Time
Session
07:55–08:00
Welcome – Rola & Spiegel
08:00–08:30
Introduction & Clinical Case – Crager
08:30–09:00
How the Blood Goes ‘Round – Miller
09:00–09:30
Andromeda 2: What Did We Learn? – Hernandez
09:30–10:00
Understanding RCT Limitations at the Bedside – Lynn
10:00–10:30
Coffee Break
10:30–11:00
Hot topic TBA!
11:00–11:30
RV at the Bedside – Siuba
11:30–12:00
“Mind of the Expert” Case Discussions – Panel
12:00–13:00
Lunch
13:00–13:45
Keynote Lecture: Changing the Oxygen Paradigm – Marino
Split Tracks – Afternoon
Respiratory Track
Time
Session
13:45–14:15
Understanding FRC – Daxon
14:15–14:45
Managing the First Hour of Severe Respiratory Failure –
14:45–15:15
Managing the Next 24 Hours of Severe Respiratory Failure – Spiegel
15:15–15:30
Coffee Break
15:30–17:00
Workshops
Extreme Hemodynamics Track
Time
Session
13:45–14:00
The RV Waveform: Clinical Applications – Denault
14:00–14:15
Understanding Afterload – Magder
14:15–14:30
Renal Hemodynamic Manipulation – Corradi
14:30–15:00
Surfing the Spectrum of Tamponade – Scott
15:00–15:30
Coffee Break
15:30–17:00
Workshops
DAY 2
Single Track – Morning (Case-Based with Workshops)
Time
Session
08:00–08:20
Case 1 – Miller & Haycock
08:20–08:40
Case 2 – Spiegel & Hockstein
08:40–09:00
Case 3 – Denault & Scott
09:00–09:20
Case 4 – Crager & TBA
09:20–10:40
Workshops & Coffee (VTI / EF / TAPSE / VExUS)
10:40–11:00
Case 5 – Kattan & TBA
11:00–11:20
Case 6 – Argaiz & TBA
11:20–11:40
Case 7 – Siuba & TBA
11:40–12:00
Case 8
12:00–12:30
Keynote: PreOx 2026 – Weingart
12:30–13:30
Lunch
Split Tracks – Afternoon
Respiratory Track
Time
Session
13:30–14:00
Navigating NIV Choices – TBA
14:00–14:20
Can I Give Inhaled Drugs to NIV Patients?
14:20–14:40
Approaching Vent Dyssynchrony – Chatterjee or Mireles-Cabodevila
14:40–15:40
When Do I Reach for Inhaled Drugs in Intubated Patients? – Daxon
15:40–16:30
Workshops & Coffee
16:30–17:00
CPAP as a Mode of Ventilation – Spiegel
17:00–17:30
Thoracic Electrical Impedance in ALI– Viau-Lapointe
Extreme Hemodynamics Track
Time
Session
13:30–14:00
The Doppler Envelope: Fundamentals – Argaiz
14:00–14:20
Resuscitating Aortic Stenosis – Augustin
14:20–14:40
The Modified Tei Index – Haycock
14:40–16:00
Workshops & Coffee
16:00–16:30
Using Cerebral Oximetry to Tailor Resuscitation – Denault
Great lecture exploring the grey zone of high risk intermediate PE. To me a very interesting area. Don’t let anyone tell you there is clear evidence on how to manage these patients – there isn’t. There’s a lot of nuance and we don’t quite yet have a grasp on how to really know who needs more aggressive management. People like Asher and those pushing the envelope with PERT teams and exploring this space will hopefully get us there.
My personal take is that the answers will be in using combined markers of congestion and forward flow (because obstructive shock both congests and restricts CO) along with some (CT?) measures of anticoagulation-responsiveness of clot burden, with dynamic cardiopulmonary reserve measures.
We’re super excited about coming to NYC, but even more about unleashing The VExUS Course 2.0, which will include the integration of venous congestion into our comprehensive hemodynamic interface framework. So participants will have an all-new version of the course, along with an introduction to the interface concept which is gaining a lot of traction, and (of course we are biased) we feel is the way forward in assessing and managing any hemodynamic issue. So this will be 4 hours of hands-on workshop and case discussion. We will also introduce participants to the use of the FlowPatch, a Doppler system for the neck veins that assesses both forward flow and congestion, and fits very well into the interface system. This course is for those who believe (as we do) that tailoring treatment to the individual patient’s pathophysiology is the way forward. Participants will leave having levelled up.
Who Should Attend?: Any clinician looking after sick patients, particularly those involved in resuscitation, congestive heart failure, and kidney injury. Learning Objectives: (1) To understand the pathophysiology behind venous congestion, (2) To be able to analyse venous congestion with bedside ultrasound, both with traditional devices and with the Flosonics FloPatch, (3) To be introduced to the Hemodynamic Interfaces and learn how to incorporate findings of venous Doppler within this framework. Course Format: flipped classroom with 4hr Pre-Course Material and 4h Hands-On Workshop. Cost: $499 Physicians/$399 Trainees. No CME.
So for the last couple of years, originally at the request of my good friend Jay Chatterjee of Riverside, California, we have been putting together a series of hemodynamic lectures, featuring several familiar H&R faculty who really love this topic, for the Riverside and Santa Cabrini critical care fellows. This past year, we basically went thru our interface model (https://pubmed.ncbi.nlm.nih.gov/40423078/), and were joined by a new group of fellows from Aurora St-Luke’s, and this year will welcome fellows from Ben Daxon’s program at Mayo.
These rounds consist of a panel discussion followed by case discussion and encourages – no – requires participation from the fellows. We’ve found that all have taken intuitively to this hemodynamics model particularly when tied to clinical cases to get some reps into the practical use.
So this year, we are planning an 8 session series, 90 minutes each, running from September to June, once a month. Fellows will have some pre-reading to do and several will be requested to present a case with challenging hemodynamics for the panel to discuss.
We would like to give other fellows the opportunity to take advantage of this unique set of discussions, and this year will add one group of fellows, so if you are a program director or involved in critical care fellowship education, reach out to hospresusconference@gmail.com and let us know. It will be first come, first served. The registration fee for a program to attend will be 499$ USD.
Unfortunately, we will not be accepting individual fellows for this as we feel that implementing an interface-based hemodynamics approach will work far better as a team than via individuals.
Everyone is welcome to watch the first iteration of these below:
FACULTY – Jay Chatterjee, Korbin Haycock, Pedro Salinas, Philippe Rola, Rory Spiegel, Ashley Miller, Sara Crager, Jon-Emile Kenny, Matt Siuba…and more.
Thanks and to the fellows, looking forward to meeting you in September for our 2025-26 series!
Here’s a throwback to #HR23 when Andre Denault first introduced the PAC with an RV port to analyze the RV waveform, which we usually only have during initial advancement of the PAC (I confess I have sometimes pulled back just to get it and look at that slope!). But these are now available! As always, invaluable hemodynamics by the grandmaster Andre!
So when we saw this study come out last week, we thought it was worth having a little chat about it, so let’s see what my usual suspects (when it comes to TCAV and, well a lot of other things…) had to say about it, and of course about TCAV in general!
So for anyone who hasn’t yet heard, #HR25 is coming up in a couple of months, and while this year, we are not specifically talking about TCAV, it will be absolutely fantastic, and there is an awesome ventilation pre-congree course, Eduardo Mireles Cabodevila’s SEVA Course, and of course Rory, Korbin and I will be more than happy to hallway talk your ear off about TCAV, so come and hang out!
Of course, for those who really want to deep-dive APRV-TCAV, our Flipping the Vent course is available online, and if you have a group/team, we can organize an online workshop as well.