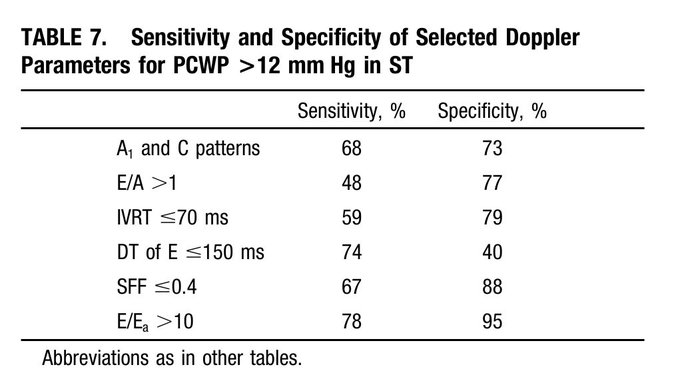
So one thing we all pretty much agree on is the importance of source control. Biliary sepsis is one of the more common causes of intra-abdominal sepsis, and among those, there is a not insignificant proportion of cases where a percutaneous drainage procedure is indicated, often related to an elevated surgical risk.
This is the case of a 90 year old man with severe aortic stenosis and a perforated cholecystitis with sepsis (AKI, delirium, coagulopathy) admitted to our ICU. Due to the aortic stenosis, surgical mortality was felt to be quite elevated, hence a percutaneous procedure was done.
I am sharing this to make the case that a percutaneous cholecystostomy is not outside the reasonable skill set of a clinician who is both POCUS competent and has solid guided procedural experience (central lines, thoracic or abdominal pigtails, etc) and in my opinion falls into that same category as pericardiocentesis. All the more so for clinicians working in community hospitals without the luxury of a 24/7 IR team, because in many cases, it is simply not reasonable to wait many hours for source control – the fact that the patient may make it alive to the next morning to have a drainage procedure is not relevant, as the ongoing sepsis over several hours may be something he or she does not always recover from in the ensuing days and is not a risk worth taking unless there is no other viable option. In our center the critical care physicians perform this intervention when IR is not available.
Here, an in-plane approach was chosen with a trans-hepatic route in order to avoid potential peritoneal spillage.
POCUS Pearls:
(1) Always visualize the guidewire inside the intended space.
(2)When dilating, make sure the proximal part of the guidewire within the target area “disappears” ultrasonographically, confirming entry of the dilator. Why? In some cases the wall may give more resistance (particularly an inflammed pericardium) and the dilator may remain outside – cannulation with the catheter will be impossible.
Procedure:
POCUS Clips
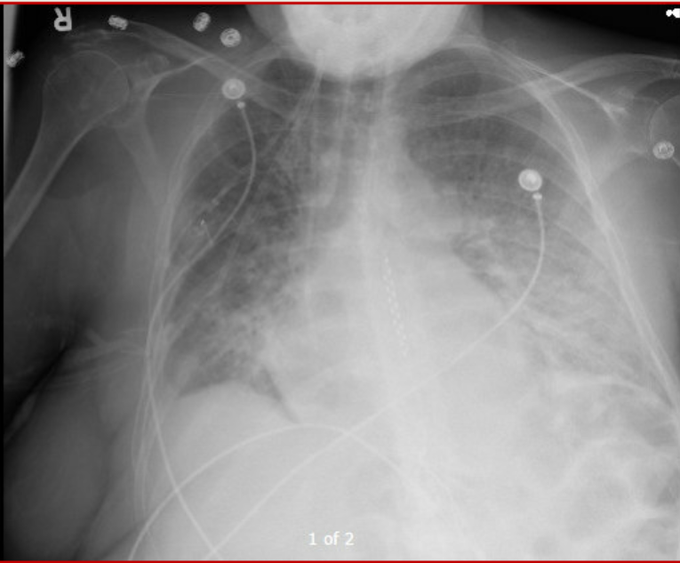
And the nasty stuff:


Some relevant articles:
https://www.ncbi.nlm.nih.gov/pubmed/12040818
https://www.ncbi.nlm.nih.gov/pubmed/29519331
Love to hear of others’ experience,
cheers
PS if anyone wants a perc chole workshop at H&R2020 , let me know!

Philippe