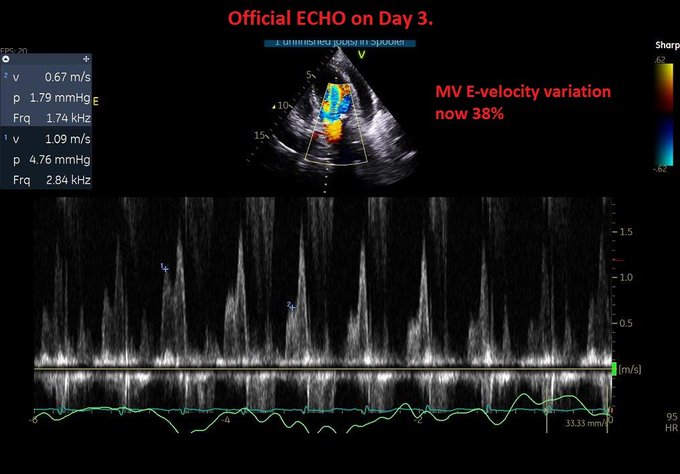
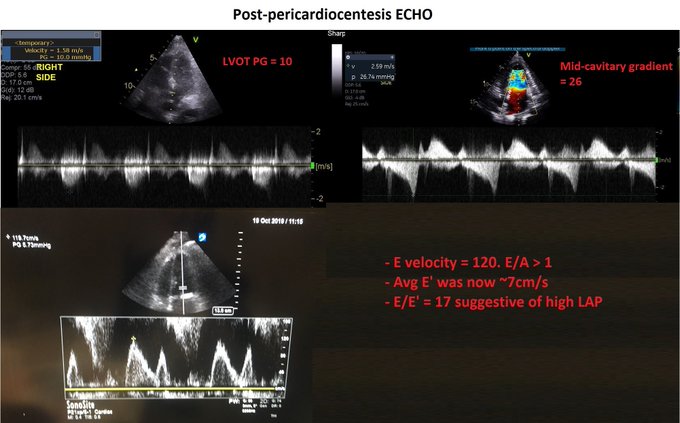
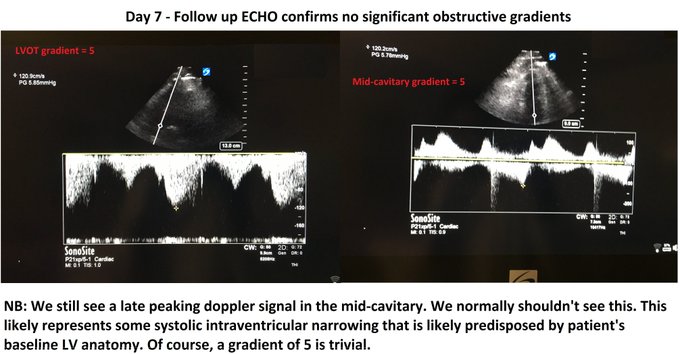
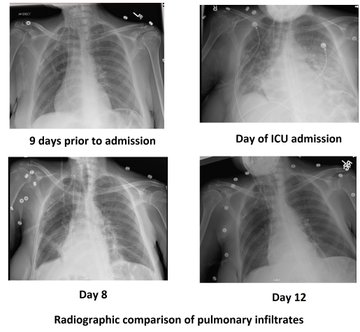
As with all continuing medical education events, this one will make you a better physician. But we know who you are: It’s 2am. Your pager is blowing up. You go to the floor with 3 simultaneously crashing patients. We’ve all been there. As a result, we’ve collectively designed this educational event to bring together an ultimate think-tank on how to improve your clinical management of all those things that make us scared at night, and even in the day… If it’s an organ that can fail, we’ve got you covered for a solid, easy, memorable approach to how to support it.
Mixed throughout the day will be cases to solidify your newly acquired clinical pearls & hands on stations with all the toys you need to stabilize your patients.
Hosted by The IBCC (Internet Book of Critical Care) co-creators Adam Thomas and Josh Farkas – also the man behind PulmCrit – this is going to be jam-packed with physiology, clinical pearls, interactive and case-based. If you take care of patients on the wards, this is one not to miss.


Co-directors: Adam Thomas & Josh Farkas.
PRELIMINARY PROGRAMME
Hypotension on the wards – Adam Thomas reviews the physiology of shock, the use of invasive & non-invasive monitoring, POCUS & “how your radiologist can help”, initial stabilization (hint, it is not just giving fluid), the hunt for & elimination of hypovolemic, cardiogenic, obstructive shock & distributive shock, as well reviewing the role of hormones and regulatory cytokines as well as how this can potentially be modulated.
Recognizing Illness at a Glance – in this interesting talk, Daniel Kaud shares his data-linkage and pattern recognition skills on common but important pathologies, to help clinicians develop rapid muscle memory and make elusive diagnoses.
Initial stabilization of respiratory failure – this can be one of the most harrowing and time-critical clinical scenarios facing the hospitalist, before the critical care team can take over or the patient can be transferred. Adam Thomas takes participants thru the identification of respiratory failure, the rule of 2s in type & treatment of respiratory failure and the right tools for the job on the wards.
Managing the Congestive Heart Failure Patient – here, Philippe Rola introduces a physiologic and POCUS-based approach to the management of the admitted CHF patient, particularly with the management of effusions and venous congestion.
Physiologic approach to Renal Failure – nephro-intensivist Sharad Patel drags the management of this common disease into this century and will share a rapid approach based on evidence, physiology and the efficiency that POCUS brings to bedside diagnosis and clinical decision-making.
The Biliary patient – whether neoplasia or lithiasis, these patients are often real puzzles. Echo? ERCP? MRCP? Drain? Stent? Fever? Jaundiced? Let’s lay down a solid base for approaching these before calling for the GI SWAT team.
Cirrhosis for the Hospitalist – in this one, hepatologist Ahn Le reviews the most pertinent pearls related to the care of patients with cirrhosis, such as managing encephalopathy, ascites, coagulopathy and more.
ID pearls – microbiologist extraordinaire Silvana Trifiro runs us through some interesting cases to make sure we don’t overlook sometimes subtle symptoms and signs of unexpected infectious diseases.
Neuro pearls – Jeff Scott shares some interesting cases highlighting key elements of the examination and management of neurological emergencies.
Wound Management – microbiologist Marc Laroche sheds some light into what is for most of us a nebulous topic, and provides a thorough, but simple and practical approach to the dressings and management of the various wounds that hospitalists come across.
Lytes, a Pot-Pourri – Josh Farkas, inventor of the “Nephron Bomb,” brings his unique, hard-hitting physiological approach to electrolyte management. This is the thinking doc’s approach, not a check-the-box one.
Clinical Cases – tying everything together, Sharad Patel, Adam Thomas, Silvana Trifiro and Josh Farkas will discuss several cases bringing together several of the key concepts and skills explored during the day.
WORKSHOPS:
- Airway: sharpen your airway skills with BVM technique, intubation, and for the bold, emergency surgical airway! Never know when you’ll need it!
- POCUS – assess the IVC and basic cardiac views for hemodynamics, assess kidneys and bladder as part of your AKI workup, assess lungs for B lines and effusions. These abilities are sine qua non for acute care at any level.
- Non-invasive Ventilation – learn how to setup and check high-flow nasal nasal cannulae, cPAP and BiPAP for your hypoxic patients.
- Pleural pigtail catheters – a simple, but important skill that should be in the skill set of all hospitalists.
- …and more on request!

We reserve the right to make the programme even more awesome by adding to or modifying the above, and promise you’ll come out of this one with a few extra notches on your belt!
But wait…only 30 spots. So don’t wait till the last minute!
click here to register!

Hope to see you there!
…and of course, if you stumbled on this, do make sure to check out the main event, H&R2020!
Adam, Josh, Philippe & The H&R2020 Team.