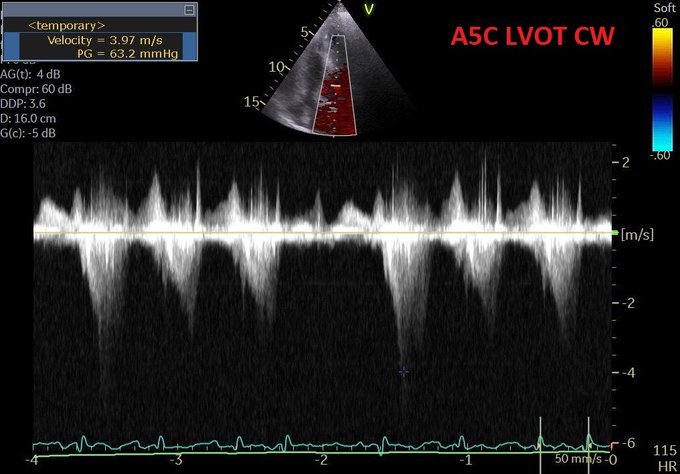
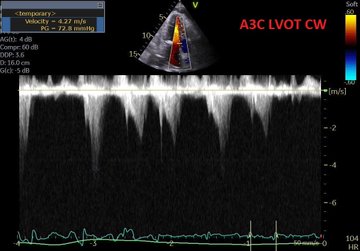
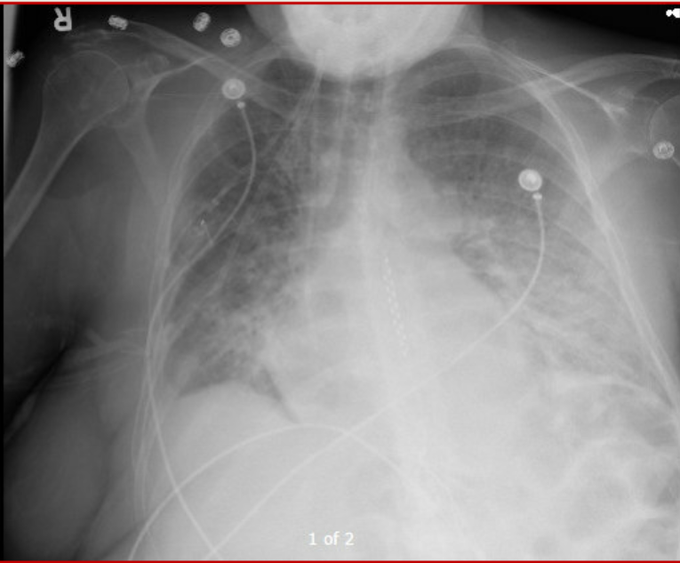
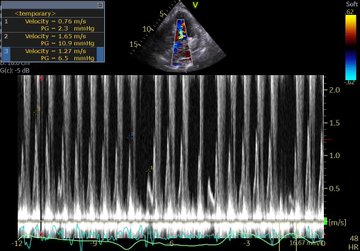
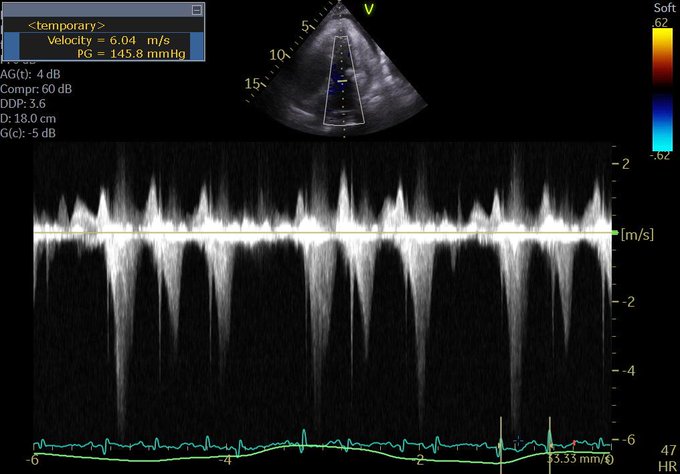
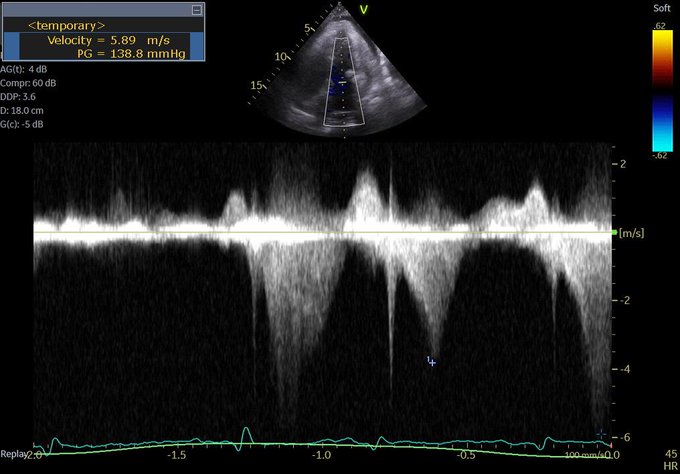
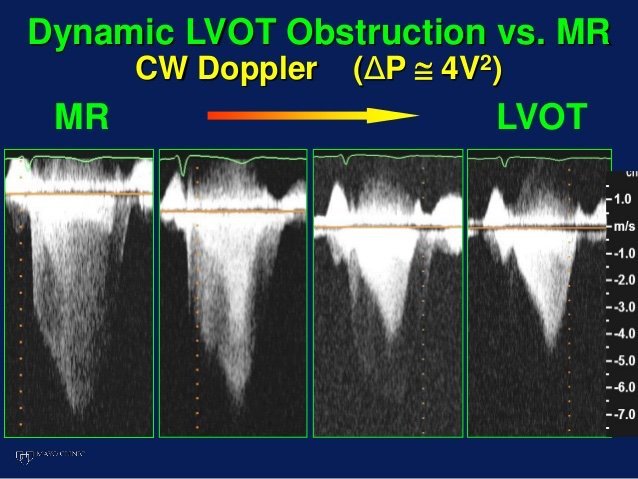
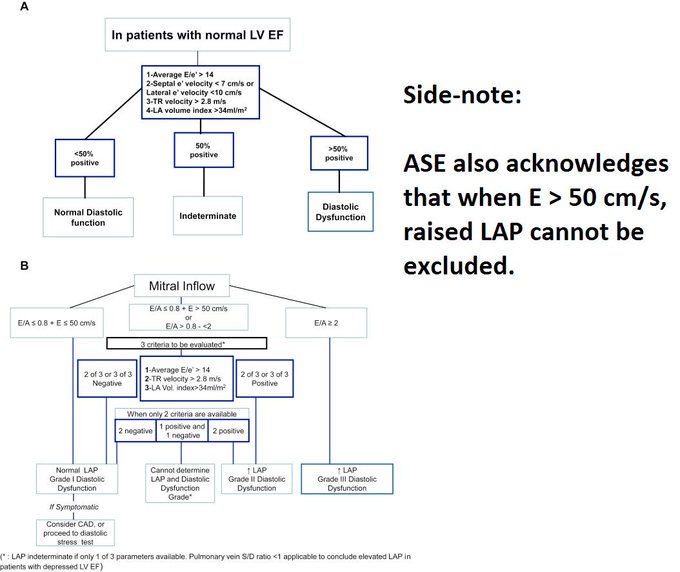
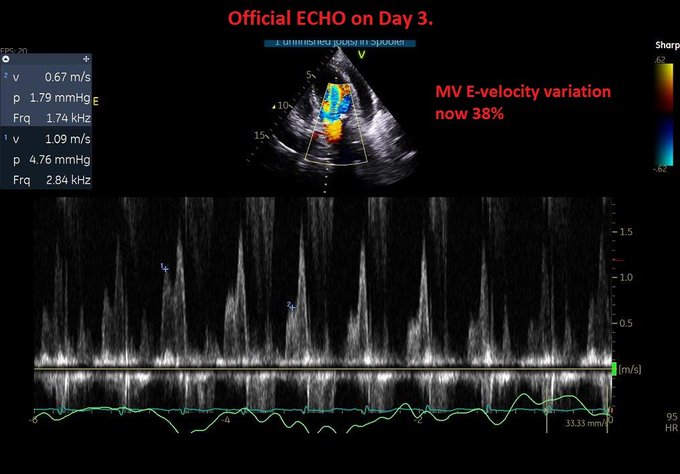
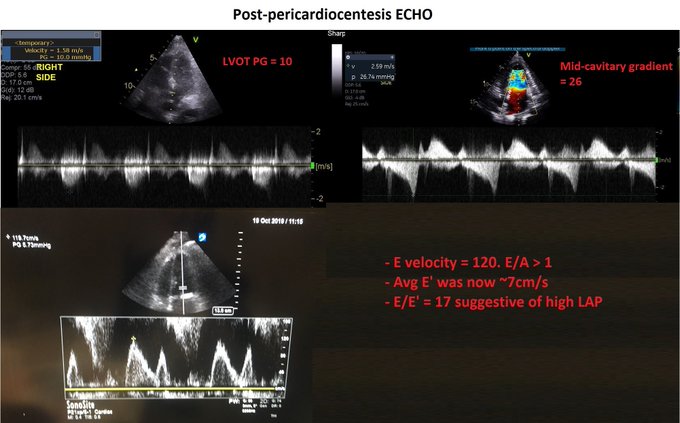
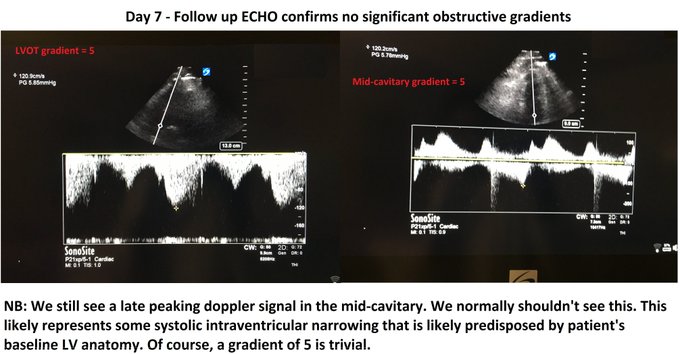
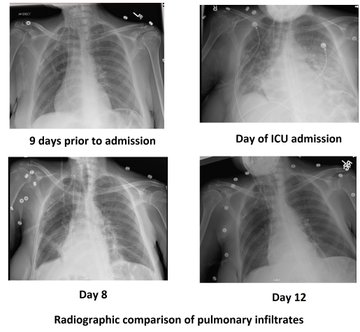
So I’m still trying to digest the RVOT Doppler physiology and working my hand at generating the best views and Doppler angles I can (See previous post on RVOT Doppler here). Not sure yet how this will fit in to my clinical practice but I think it’s worth shining a light into this murky pseudo-science of resuscitation. These guys are helping define its potential use… Naturally, this is bleeding-edge stuff. Use it to try to understand what’s going on with your patient’s physiology, don’t use this on board exams! My comments in bold.
Now for the big guns…

Jon-Emile Kenny (@heart_lung), pulmccm.org, heart_lung.org
Hey Guys – great discussion as always!
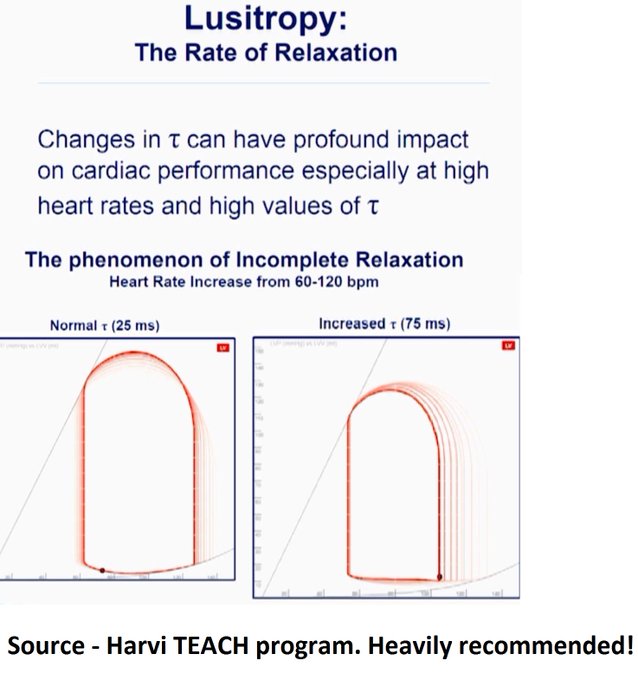
One thing that I find confusing on this topic, and is helpful – i think – when scrutinizing the literature, is the difference between ‘impedance’ and ‘resistance.’ Elevated vascular ‘resistance’ is often used too broadly; for example, true/pure WHO II pulmonary venous hypertension [say from acute left atrial pressure hypertension, but before chronic, compensatory pulmonary arterial changes] is actually typified by a *decrease* in resistance, but an increase in *impedance.* To make things more confusing, acute left atrial hypertension will often display a high “resistance” mathematically … even though, the true resistance can be low. What am i saying? if you imagine an acute increase in the left atrial pressure, the pulmonary venous beds and pulmonary vascular beds “recruit and dilate” backwards [why we see cephalization on the CXR] typically from the bottom to the top of the lungs up the hydrostatic gradient. Recruitment and dilation actually *increases* the cross-sectional radius/area of the vascular beds — a true decrease in resistance [Poiseuille what?]! But, as these vascular beds are engorged, they reach that infamous, hockey-stick-shaped compliance curve point [go leafs go!], where the vessels become really stiff … that is, the compliance falls such that each ejection the RV throws into this dilated circulation, the pressure rises dramatically [especially the systolic pulmonary pressure] …
This I think is a key concept to understand and keep in mind when analyzing the venous system. The physical characteristics are more akin to a floppy plastic bag or balloon, with little rise in pressure until a certain point, then a sharp one – Jon’s “hockey stick.” It was Jon who made me realize that, with exposure to chronically elevated right atrial pressures, one could have a very big IVC (say 25-30mm, but in fact a low CVP, whereas in normal IVCs exposed to normal CVPs, that sharp rise in pressure probably occurs somewhere around 20mm. Hence, the + value we use in the PAP calculation using TR Vmax for the RAP may be very inaccurate in chronically elevated PAP… Food for thought.
Thus, the calculated pressure gradient rises and and the calculated resistance falls, but what has actually happened is that compliance has fallen, not “resistance”. More broadly, the term “impedance” is composed of compliance, resistance and something called the characteristic impedance [the Windkessels!]. Typically what abnormal RV Doppler shows you is that *impedance* has risen. At the end, you are often still left with the why? Impedance can rise when “true resistance “falls, but compliance also falls [as above] – yet the calculated RVSP/regurgitant jet will also rise. The linked papers are fantastic, but they both excluded patients with left heart disease, so you can be more confident that the RVOT abnormalities seen are related to true ‘pre-capillary’ problems. I’d be willing to bet [and if there’s data, i’d be interested to read it] that patients with pure WHO II pulmonary venous hypertension have very similar abnormalities on the right side. The key means to distinguish – as Korbin talks about – is really looking at the left heart [E/e’] and clinical context to get a better sense of what’s going on.
What would also be interesting would be to look at acutely “decompensated” true left heart disease in volume overload and correlated with RVOT morphology and great vein Doppler velocimetry. My guess is that as you decongest the pulmonary veins [increase their compliance] that the RVOT envelope “pulsatility” goes away [the RV ejection envelope appears more rounded] as does the venous pulsatility in the great veins and intra-renal veins! It’s all about energy transfer … moving away from excessive potential energy trapped in distensible structures [i.e. congestion] to kinetic energy [normal, forward blood flow]
Jon.

Korbin (@khaycock2)
Thanks for the reply Jon-Emile, as usual you bring an incredible amount of intelligent well thought out points.
As you mentioned, afterload is much better described in terms of the 3-element Windkessel model as resistance is only one component of said model (the other factors being vascular compliance and characteristic impedance). Please correct me if I’m wrong, but I believe that the most practical and easiest way to non-invasively determine arterial load is to calculate the Ea (formula: (SBP*0.9)/SV). This would include all of the factors that determine afterloading conditions instead of simply using resistance as it is only one of those factors.
Clinically speaking, I think it is important to address why afterloading conditions are abnormal when we come across undifferentiated pulmonary HTN in the acute setting. Practically in my mind, this is simply finding if the pHTN is due to post-capillary “back pressure” from elevated left atrial pressures or due to elevated pre-capillary pulmonary vascular resistance (or could be some combination of both of course). Both of these conditions can cause elevated pulmonary artery pressures, as you have pointed out, and there are a few other contributors to the afterload as well that we are ignoring (or else we’d blissfully nerd out all day and forget to take care of the patients).
I agree with this concept. This is what may direct me to use pulmonary vasodilators, whether inhaled or even the choice of milrinone or vasopressin (not a vasodilator per se but a non-pulmonary vasoconstrictor). If all the pulmonary hypertension is post-capillary, there would be little or no benefit. This important decision point is what prompts me to look into this whole right-sided Doppler thing… Let’s see what else Korbin has to add!
So how can find out the cause(s) of the elevated PAP? Is it resistance or back pressure from the left atrium? This is essentially the topic of the post. Because PVR=(mPAP-LAP)/CO, it has been suggested that the TR gradient can be a surrogate for the mPAP-LAP and RVOT VTI be a surrogate for CO. Thus if the ratio is high, we can assume that a significant component of the pHTN is due to resistance in addition to or to the exclusion of the contribution of LAP. You have rightly questioned and very well explained why you wonder if these are valid assumptions that translate to the finding the clinical causes of pHTN.
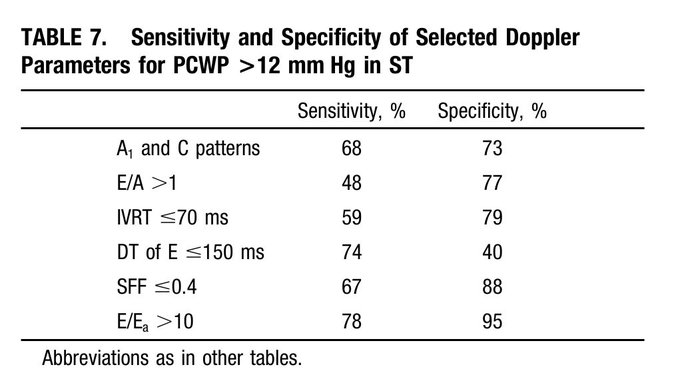
You pointed out that the cited papers in the post excluded patients with LV failure, thus bringing into question if the TR/VTI ratio methods and their permutations are actually detecting PVR as the primary etiology of the pHTN or are corrupted by elevations in LAP. Here are 3 papers that included a significant number of patients with pHTN and elevated PCWPs as measured by RHC that show that the TR/VTI methods do seem to work to detect PVR elevations themselves even if the LAP are high:
1) Am J Cardiol. 2013 September 15; 112(6): 873–882. doi:10.1016/j.amjcard.2013.05.016.
2) J Am Soc Echocardiogr 2013;26:1170-7.
3) J Am Coll Cardiol 2003;41:1021–7.
Somewhere in my files I have a study that shows that the mid systolic notch is fairly specific for high PVR and independent of LAP as well. but apologies, I’d have to look for it.
As I might have mentioned in the audio portion of the post (I can’t remember), there is a second method to flesh out PVR from LAP causes of pHTN. First, you need to find a good estimation of the LAP. ECHO has multiple ways of various accuracies to get a number. The formulas are listed above. I don’t believe any of them are validated in acutely sick patients though. Once you have a LAP number, turn your attention to the pulmonary valve regurgitant jet which will almost always be there if there’s pHTN. The wave form is sort of down-sloping trapezoidal lasting through diastole. The velocity at end-diastole can be squared, multiplied by 4, then added to the RAP to give you the end-diastolic PAP. This is normally < 6 mmHg higher than the LAP pressure measurement, if it is a bit more higher, there likely is increased PVR. This is the same principle used in a RHC, where the inflation of the balloon stops flow and therefore eliminates resistance so that the PCWP can be measured and differentiated from the dPAP. The problem with this method is that it doesn’t work as well as the TR/VTI methods
I really enjoyed your thoughts about how Doppler waveform patterns may be affected once compliance limits have been reached, and I’m sure there is something to this that is real as well no doubt! I thought it might be helpful to provide you with the additional studies that included the patients with high LAP, and do a bit of re-explaining/restating your points to anyone new to this stuff.
Thanks again Jon!
Jon replies:
Hey Korbin – thanks for the references – I’ll dig into them. My main concern is that the mPAP-LAP will disproportionately rise (mostly because the sPAP disproportionately rises) when the left atrial pressure is high … that is when it’s actually not a “resistance” problem but rather a back pressure problem, the mathematical resistance is high. As you mention, this is why there’s a push to move away from “PVR” with RHC and more towards the dPAP-PCWP gradient which should be less than 6 mmHg. I made a cartoon describing this in an old post (https://pulmccm.org/critical-care-review/icu-physiology-1000-words-folly-pulmonary-vascular-resistance/). Thanks for these references, I’ll read them and see if they make sense from the framework I’ve adopted – which is entirely stolen from this great article
Naeije, R., et al., The transpulmonary pressure gradient for the diagnosis of pulmonary vascular disease. Eur Respir J, 2013. 41(1): p. 217-23.
Maybe Phil should do a point-counterpoint podcast where Rory comes in at the end and shakes his head because nothing really matters in the end.

“Nihilism rules…”
Jon
Korbin:
Thanks Jon, I would like to see what you think. Thanks back at you for the reference you mentioned in your reply. And you’re hilarious!
Jon replies:
I had a read of the references that you provided, thank you. I think my concerns still apply, however. My main concern is what is being used as the gold standard for ‘pulmonary vascular resistance.’ An elevated calculated pulmonary vascular resistance (e.g. in WU) doesn’t actually tell you where the pathology is. the assumption is that an elevated calculated pulmonary vascular resistance is caused by a high pre-capillary resistance in the pulmonary circulation, but this isn’t necessarily true. as i showed in that post that i linked to (https://pulmccm.org/critical-care-review/icu-physiology-1000-words-folly-pulmonary-vascular-resistance/) … if one were to acutely cross-clamp the descending aorta, below the diaphragm, the calculated pulmonary vascular resistance would rise, even though the pathology is totally outside of the thorax!! i have no doubt that the TRV / RVOT-VTI would also rise in that very same patient with the cross-clamped descending aorta such that the good correlation between the calculated ‘pulmonary vascular resistance’ and the TRV / RVOT-VTI is maintained – but the pathology is in the abdomen – not the pulmonary vascular tree! So many exclamation marks; but i’m not yelling. In a hypothetical patient with a cross-clamped descending aorta, one might be tricked into giving a pulmonary vasodilator — but that would be the absolute wrong thing to do, even though the calculated pulmonary vascular resistance is high. The treatment is to afterload reduce the struggling LV (remove the cross clamp) — which would then lower the calculated “pulmonary” vascular resistance and the TRV / RVOT-VTI.
the problem in reasoning lies in what happens with the left atrial pressure rises (as would happen if one acutely cross-clamped the descending aorta). it is assumed that as the LAP rises that the mPAP – LAP gradient stays the same or rises in proportion. but what happens when the LAP rises is that the mPAP rises disproportionately because of pulmonary vascular engorgement/stiffening (in fact, the pulmonary vascular resistance has fallen because of recruitment and dilation of the pulmonary tree). what *does* rise in proportion is the dPAP – LAP gradient [should stay below 7 mmHg]. i strongly suspect that the ability of the TTE to detect/calculate the dPAP – LAP gradient is not yet refined enough because there is a lot of supposition and inference when making dPAP and LAP measurements with pulsed wave Doppler.
alas, with either an elevated TRV / RVOT-VTI (or calculated pulmonary vascular resistance from a RHC), one still doesn’t know if it’s purely a left-sided problem (e.g. purely elevated LV afterload) – which could seriously alter management. to know that, i think that a full interrogation of the left heart and pulmonary veins must be done before knowing exactly what an elevated TRV / RVOT-VTI specifically identifies. in addition to that vascular resistance post above, i dug into some more of this in a discussion on the SIOVAC trial a while back (https://pulmccm.org/randomized-controlled-trials/choose-wisely-avoid-sildenafil-pulmonary-hypertension-corrected-left-heart-valvular-disease-siovac-trial/) – which, in my opinion, should never have passed ethics.
Jon
So this is really fascinating stuff. I must admit both Korbin and Jon make excellent points, and for now am not sure if and how to use RV Doppler in clinical decision-making, but until then will be sure to polish up these skills so that they are ready for prime time, and use them in observation of physiology in my shock patients. We’ll see what conclusions I draw.
cheers
Philippe