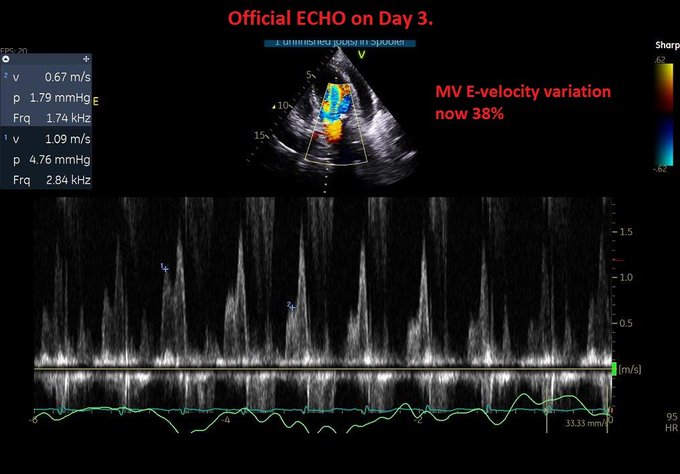
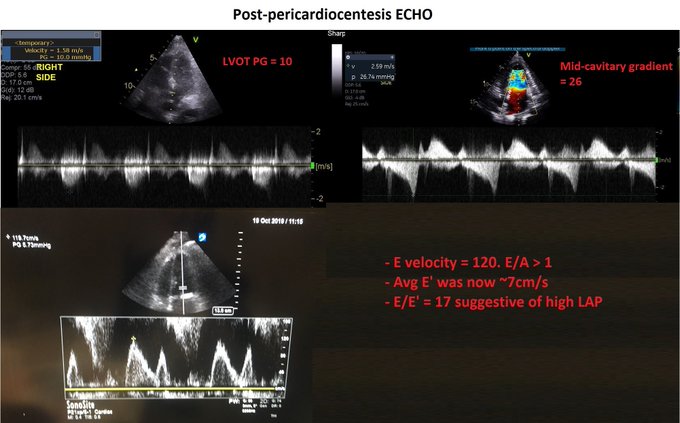
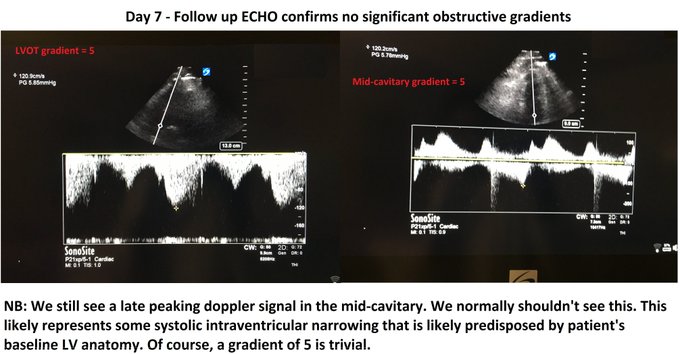
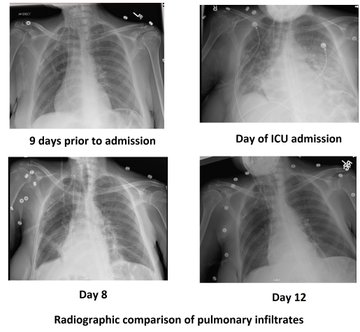
A few words about it…
HR2025 is about putting it all together. We’ve been talking about the venous side for a long time and it has been fantastic seeing how much it has taken off – at least in the #medtwitter #foamed and PubMed community. There’s enough data out there to show a real clinical utility of consciously examining the venous compartment. This applies to any hospitalized patients, whether in the ED, the wards or the ICU, so that’s why this year, the H and the R will spend the first day together doing all things VExUS and venous congestion, along with the corresponding workshops, again so that participants can leave with some actionable knowledge and skills. Whether you’re a beginner at this or a seasoned veteran, there should be something to learn.
On Day 2, we’ll divide into two tracks. The R side will deep-dive bedside hemodynamics and our 4-interface model of shock management. Of course there will also be some pearls and late-breakers as this is still a year away and there will undoubtedly be some really interesting things cooked up by the crew by then!
Meanwhile on the H side, Katie and Gigi are thrilled to have put together a top-notch lineup of speakers to teach everyone how to become Inpatient Medicine Jedis. We have some big names in attendance this year, including both new and familiar faces – The Clinical Problem Solvers themselves will take us through some challenging cases; Frederik Verbrugge will drop diuretic truth-bombs; the ever-fabulous @NephroPOCUS will discuss a Bedside Approach to AKI; and Allison Bond will share the Top 10 Infectious Disease Mistakes on the ward, among many others! Day 2 Workshops will be focused on high-yield test interpretation: think EKGs, PFTs and sleep studies, urine microscopy, peripheral blood films, and more!
https://player.vimeo.com/video/1022725950?h=825106f34a&badge=0&autopause=0&player_id=0&app_id=58479
And as always, the ethos of H&R is about putting together physiological clinicians who love to both push the envelope and share their knowledge and experience. The energy that comes out of this is really quite unique, and the sheer number of successful collaborations that have stemmed from it since 2018 is really impressive. The unplanned, unscheduled small group discussions are the true gems of this conference… Many of the usual suspects will be there, and as always some new additions to the H&R family!
Who? I can’t say enough about the H&R crew. Brilliant, open minded, eager to both learn and teach, no large fragile egos here. So expect to learn from and hang with Sara Crager, Korbin Haycock, Rory Spiegel, Matt Siuba, Eduardo Argaiz, Gigi Liu, Ross Prager, Frederic Verbrugge, Vimal Bhardwaj, Glenn Hernandez, Andre Denault, Jon-Emile Kenny, Ashley Miller, Segun Olusanya, Max Hockstein, Ben Daxon, Abhilash Koratala, as well as some new faces to the live event, very much looking forward to meeting Trina Augustin from Mayo as well as another critical care icon, Professor Jan Bakker, and I’m happy to announce the return of the true father of POCUS, Daniel Lichtenstein, and we will even have the amazing opportunity to (virtually) hear from Dr. Geoff Parkin, one of the pillars of hemodynamic physiology! There are a few more to confirm and we promise they will bring the same enthusiasm and unique experience to the event!
So the most important thing for you to do is to mark your calendars and make sure you don’t miss being a part of it! Bookmark this page as the registration link (november 1) will appear as well as developing programme information! Note that registration fees will be in USD given the international nature of the conference.
Montreal, May 21-24. Core conference May 22-23, Pre and Post-courses May 21 and 24.
Pre/Post congress preliminary courses
- BJJ or Self-Defense for Humans & Health Care Workers (May 21st pm)
- The VExUS Course (May 21st am)
- ArrestTEE Sim Cases (May 21st)
- The Great Presenter by Marco Garrone (May 21st am)
- Sauv Life (eCPR) by Paris’ Lionel Lamhaut! (May 24th)
- Bedside EEG for EDCritters? (TBA)
- SEVA Ventilator Course by The Cleveland Clinic’s Eduardo Mireles-Cabodevila! (May 24th)
- ResuscitativeTEE Workshop by Felipe Teran (May 24)
for more on these see https://thinkingcriticalcare.com/2024/10/26/hr2025-pre-and-post-courses-hr25/
if there’s a course you want, go ahead and get in touch with us! hospresusconference@gmail.com or via twitter with #HR25 tag.
Schedule
Day 1 – May 22
| Time | Talk/Workshops | Faculty |
| 0800-0815 | The Concept of Fluid Tolerance | Rory Spiegel |
| 0815-0845 | Bedside Cheat Code – Femoral Doppler | Vimal Bhardwaj and Andre Denault |
| 0845-0915 | An update on the Hepatic Vein | Eduardo Kattan |
| 0915-0945 | Understanding Portal Vein Doppler | William Beaubien-Souligny |
| 0945-1015 | Coffee Break | |
| 1015-1045 | Renal Venous Doppler – RVSI and VExUS | Eduardo Argaiz (virtual) |
| 1045-1115 | eCPR and Organ Donation | Lionel Lamhaut |
| 1115-1215 | Workshops:Mastering the IVC assessment Hepatic and Portal vein assessments Intrarenal hemodynamics Femoral vein How I assess congestion at the bedside (Virtual workshop) | Rory Spiegel, Juliana Kan and Audrey Lacasse Eduardo Kattan and William Beaubien-Souligny Abhilash Koratala and Korbin Haycock Vimal Bhardwaj and Andre Denault Ross Prager |
| 1215-1300 | Lunch | |
| 1300-1330 | Keynote Lecture – The Evolution of Venous Congestion Assessment | Andre Denault |
| 1330-1400 | Pulmonary congestion: Mastering lung US | Daniel Lichtenstein |
| 1400-1430 | Practical Diastology: Will my Pt get Pulmonary Edema? | Frederik Verbrugge |
| 1430-1530 | Workshops:Diastology, E/e’ and LA size CVP using Jugular POCUS (Virtual and in person workshop) Lung US RV assessment (TAPSE, S’, RVH, PASP) Beside Cerebral Oximetry in congestion | Frederik Verbrugge and Max Hockstein Jon Emilie Kenny and Korbin Haycock Daniel Lichtenstein and Marco Garrone Matt Siuba, Andre Denault, Juliana Kan and Audrey Lacasse Phil Rola and Masimo |
| 1530-1545 | Coffee break | |
| 1545-1615 | Fluid Tolerance in the ED – Is It Pertinent? | Marco Garrone |
| 1615-1645 | Minute Ventilation: Physical Exam Hack? | Rory Spiegel |
| 1645-1715 | Clinical Cases: Congestion | Abhilash Koratala |
Day 2 – May 23
| Time | H side talks | Faculty | R side talks | Faculty |
|---|---|---|---|---|
| 0800-0830 | Clinical Problem Solvers: Challenging Cases | Reza Manesh and Rabih Geha | Intro to the circuit & interfaces | Sara Crager |
| 0830-0900 | Clinical Problem Solvers: Challenging Cases | Reza Manesh and Rabih Geha | 0830-0850: What is Coupling? | Jon Emilie Kenny |
| 0900-0930 | Immune Checkpoint Inhibitor Toxicity 0930-0940: Buffer | Laura Cappelli | 0850-0910: Understanding MSFP 0910-0940: Resurrecting Blood Pressure. Curious Cases of B12 for Vasoplegia | Ashley Miller and Korbin Haycock Ben Daxon |
| 0940-1040 | Workshops:Approach to acid-base problems (Virtual and in person workshop) | Rory Spiegel | Workshops:LVOT VTI Capillary refill time POCUS AMA Hemodynamic Pearls Part 1 | Sara Crager, Trina Augustin and Jeff Scott Glenn Hernandez and Eduardo Kattan Jay Chatterjee, Jon Emilie Kenny and Marco Garrone Ashley Miller |
| 1040-1110 | Coffee break | |||
| 1110-1140 | Oncologic Emergencies | Aditi Singh | Interface 1: LV-VA coupling & How I Measure it | Max Hockstein |
| 1140-1210 | Bedside Approach to AKI | Abhilash Koratala | Interface 2: Macro-Micro | Glenn Hernandez |
| 1210-1300 | Lunch | |||
| 1300-1330 | Keynote Lecture – “Fill him up to his eyeballs.” | Jan Bakker | ||
| 1330-1400 | Acute decompensated heart failure | Frederik Verbrugge | Interface 3: Capillary-Venular | Eduardo Kattan |
| 1400-1430 | High output heart failure | Eduardo Argaiz – (virtual) | Interface 4: RV to PA | Matt Siuba |
| 1430-1530 | Workshops: Urine electrolytes and microscopy POCUS AMA | Abhilash Koratala Daniel Lichtenstein and Sara Crager | Workshops: POCUS RV-PA coupling PA catheter (3 simulators) Hemodynamic Pearls Part 2 (Virtual and in person workshop) | Matt Siuba and Korbin Haycock Katrina Augustin, Jeff Scott and Andre Denault Ashley Miller |
| 1530-1545 | Coffee break | |||
| 1545-1615 | Management of Non-Insulin medications in the hospitalized patient | Elias Spanakis | Can Starling and Guyton Collaborate at the Bedside? | Geoff Parkin and Segun Olusanya (virtual) |
| 1615-1645 | Next level BiPAP and CPAP | Segun Olysanya (virtual) | Impella in Cardiogenic Shock – What Every ER and ICU Doc oughtta know! | Katrina Augustin |
| 1645-1715 | Top 10 Infectious Disease Mistakes on the Wards | Allison Bond | Putting it All Together | Rory Spiegel |
Registration – https://ccusinstitute.wixsite.com/ccus/events/hr2025-fluid-tolerance-all-things-vexus-shock-hemodynamics
There will be a number of different registration options so please select carefully. Please note that for online-only registrants, we will follow a fee scale using the World Bank country classification system, so click the link below if you’re not sure what applies to you (this applies to where you live/work, not your country of origin).
Scientific Committee – Dr. Philippe St-Arnaud (Santa Cabrini Hospital, Montreal), Dr. Benjamin Daxon (Mayo Clinic, Minnesota), Dr. Rory Spiegel (Medstar Health, Washington, DC), Dr. Katie Wiskar, (University of British Columbia).

Cancellation policy: Cancellations until April 15th will be accepted and refunded minus a 5% administrative fee. Cancellations until May 1st st will receive 50% refund, and after that date no cancellations will be possible.
Hotel Information: as the hospital is in a residential area there really isn’t very much around. We suggest finding a hotel on the east side of downtown Montreal (east of University ave) or in Old Montreal, for the enjoyment of the city after the conference, especially if travelling with companions. We have found that better deals can be had using websites rather than for us to secure a conference rate (usually +30%). Priceline/expedia/booking.com are fairly reasonable. The hospital is about 20 minutes by uber/cab from these areas.
Thanks to our Sponsors!!!!!