So a lot of people have been asking for a VExUS tutorial, and since our paper was just accepted, I figured it’s a pretty good time to do it! Thanks to Dr. Ian Ajmo of FrancoFoam fame who put on his Hollywood director’s hat and filmed it!
Here is the classification that has been validated:
I’m attaching our chapter on venous congestion below as well.
Of course, Andre Denault, William Beaubien Souligny, Rory Spiegel, Korbin Haycock and myself will be running VExUS workshops at H&R2020. There aren’t many spots left!
So anyone who knows Korbin (@khaycock2) realizes he is a true trailblazer in the ED, essentially doing cutting edge critical care from the get go in his shock patients. In my mind this should be the goal for any critically ill patients, that they get the highest level care right at entry and for however long they may be staying in the ED until they get to the ICU.
So today, I was really happy to corner Korbin lounging somewhere in sunny California (as 6 inches of snow come down hard in Montreal) to tell me how he is using this technology in his resus patients.
So this has got me interested in using this technology. I see it as an early warning signal that your patient may be less fluid tolerant than you may think, and that the signs of pulmonary fluid intolerance I use (oxygen requirement, appearance of B lines (FALLS Protocol-style), etc…) have yet to manifest.
So I’m looking forward to hearing Korbin explain this further (during H&R2020!) and in actual cases where the change in management is clear.
So #MedTwitter is truly an incredible forum for case discussion, where you get to exchange with literally some of the best medical minds on the planet who often also happen to be front-line clinicians in the nitty-gritty therapeutic decision-making. Here’s a discussion which I think was great. Recently, Dr. Thind has been generating some great cases and hemodynamic discussions. I thought this one was worth highlighting!
Dr Thind is an internist and currently Critical Care Hospitalist (and upcoming ICU fellow) at the Cleveland Clinic, and tweets out some great #FOAM from @Thind888 on twitter.
Case:
OK, let’s give this a shot. Here’s a ‘hemodynamics special’. Saw this case a couple weeks ago. A lot of decision making was based on educated guesses so it should be a good one for discussion. – 51 yo woman being worked up on the floor for chronic diarrhea, moved to ICU for hypoxia.
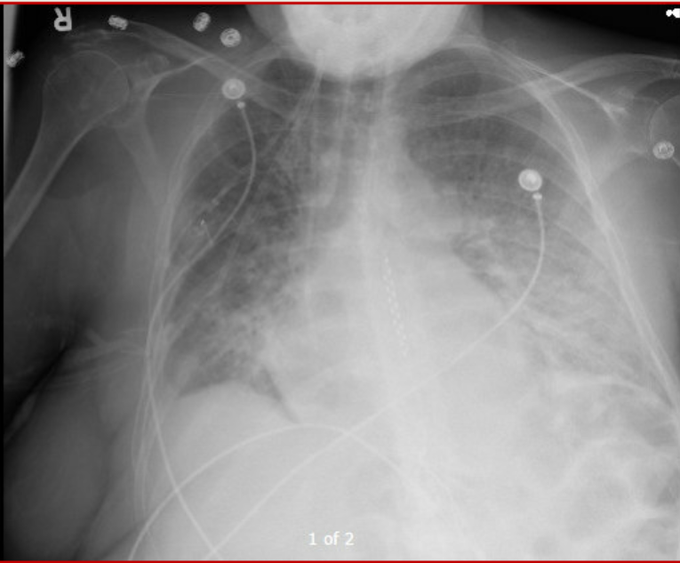
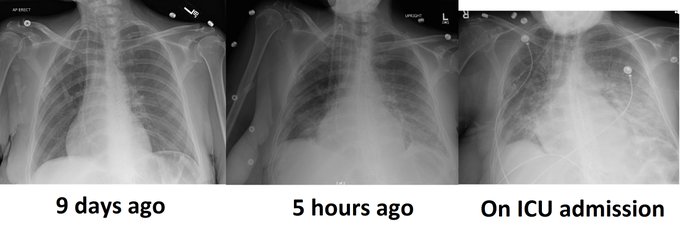
Dyspnea progressed over few hours. Vitals significant for tachycardia (140s) and hypotension (MAP in low 60s). On arrival, SBP 60s – improved with fluid bolus. CXR attached. Patient has H/O of pericardial effusion for several months that has been managed conservatively.
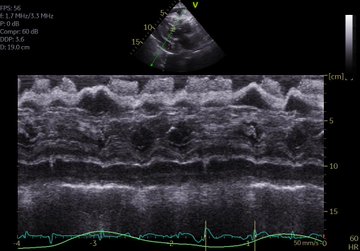
The patient has an official ECHO performed on arrival in ICU (images attached). IVC difficult to assess but about 2cm without collapse. Lung US – diffuse B lines.
OK so right there a flag goes up for me. A plethoric IVC means something is wrong. Sounds too vague maybe, but you need to find the reason for this, as it likely has therapeutic implications. Let’s see what comes up.
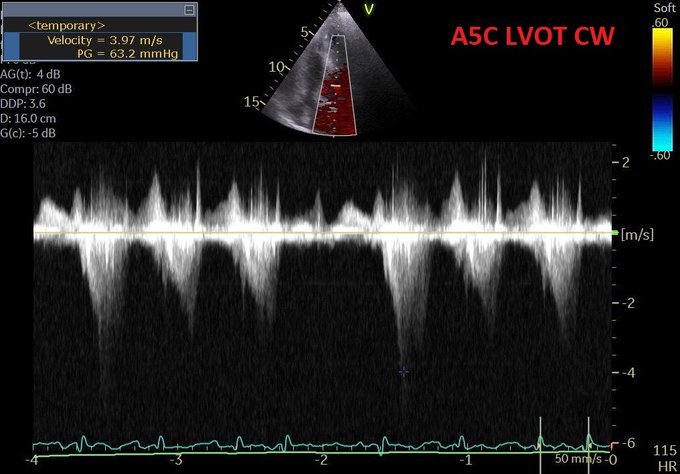
Modifed A5C.
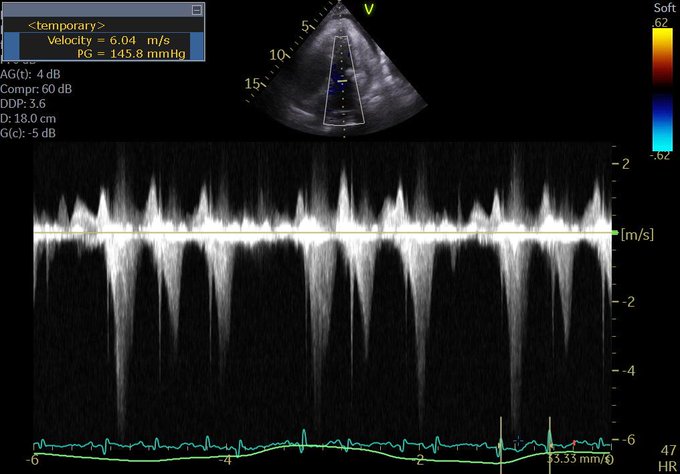
LVOT doppler
CXR
Pressing questions –
(i) Is it hydrostatic or increased permeability pulmonary edema?
(ii) Fluids, diuresis, or none?
(iii) Would CPAP help?
(iv) Drain the pericardial effusion?
(v) What about that LVOT doppler?
Mitral inflow velocities and TDI attached. M-mode through PLAX almost uninterpretable. Lung infiltrates are new so less likely lymphangitic carninomatosis. Note: ScVo2 = 40s. Another Q to ponder on –
(vi) Is tamponade typically associated with hydrostatic pulmonary edema?
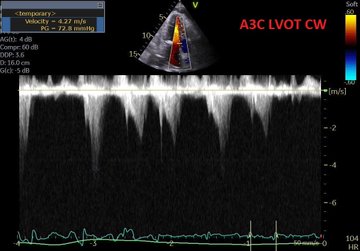
Perhaps this slowed up (0.5x) A3C loop will help with that LVOT doppler!
Great discussion as expected. Lets discuss:
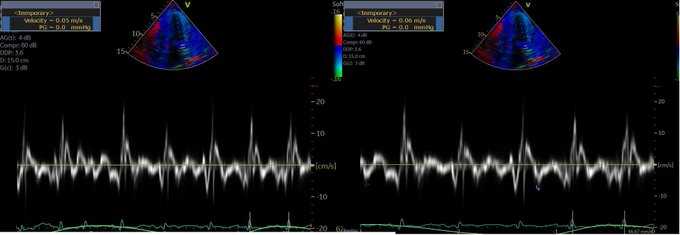
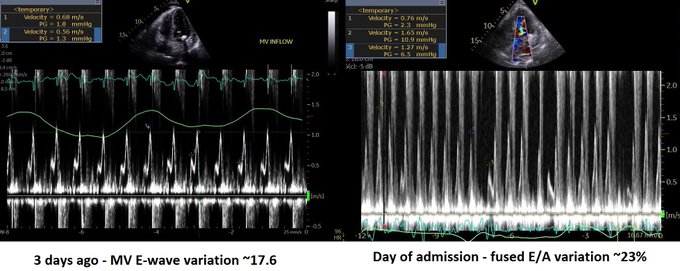
Q4. Is it tamponade? – This is not a slam dunk. Chamber collapse can sometimes be controversial. In these situations I try my best to get MV E-wave variation. I think our tech got a decent signal. But note these are fused E/A waves.
The first thing I look at to screen for tamponade is the IVC. Tamponade is an obstructive form of shock, dependant on the intrapericardial pressure exceeding the right atrial pressure. If it does, unless respiratory efforts are extreme, the IVC should become plethoric. Hence, the absence of such would make the effusion – given the current RA pressure – NOT tamponade. Yet again, another point scored by the IVC for usefulness.
Although I don’t see why we can’t use fused waves for this purpose (couldn’t find anything on it in the literature). Note that in spite of the cardiac motion, the mitral inflow variation is <25% (~23%). It’s close though, and certainly seems to have increased from 3 days ago.
The cardiologist (understandably) was non-committal and read it as “possible early tamponade”.
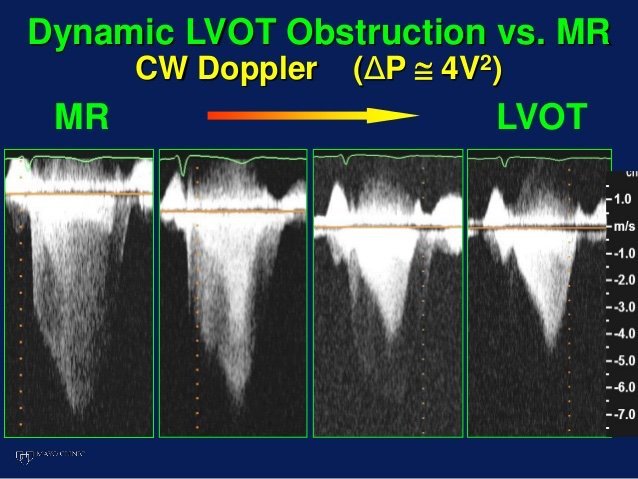
Q5. What about LVOT doppler? A good M-mode could not be obtained but the A3C in 6/ shows SAM. The report mentioned “chordal SAM” but I think you can clearly see “valvular SAM” too.
Chordal SAM is SAM of the chordal apparatus (you could see it bumping against the septum in 6/). It is (typically) NOT hemodynamically significant (PMID: 27241937). – When we see mitral SAM, it is important to quantify its hemodynamic effects – with LVOT peak gradient via CW.
In HOCM, DLVOTO is defined by an LVOT gradient of >30; >50 is considered severe. Our patient had a gradient of ~70. Although classically a/w HCM, SAM can be seen in anyone with thick, hypercontractile, underfilled LV. Tachycardia further hampers LV filling (PMID: 27726435).
Mitral SAM is often a/w MR – this acute MR can cause flash pulmonary edema. These patients may actually need fluids (to help with SAM) to fix there hydrostatic pulmonary edema!! (PMID: 20661209). However, our patient only had trace MR (you could see it in 1-2 CD frames).
Working theory (similar to Lars) – Chronic stable pericardial effusion –> diarrhea (pt had 15 BMs the day before the admission) –> reduced venous return –> brought the patient at the verge of low-pressure tamponade (PMID: 16923755) –> further reduction in LV filling —> reduced stroke volume –> adrenergic drive causing tachycardia and increased inotropy –> all factors culminating in mitral SAM and DLVOTO.
This also explains the low ScVO2. Note – CPAP would further reduce venous return (Q3) so wouldn’t help, may hurt.
Now the most important Qs: why pulmonary edema and what to do about it (Q1 and 2). As tamponade causes impedance to venous return, it is not typically associated with high LAP and hydrostatic pulmonary edema (Q6).
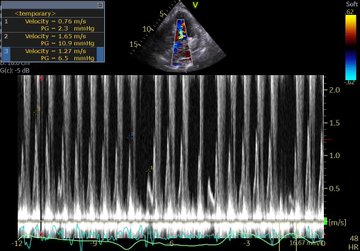
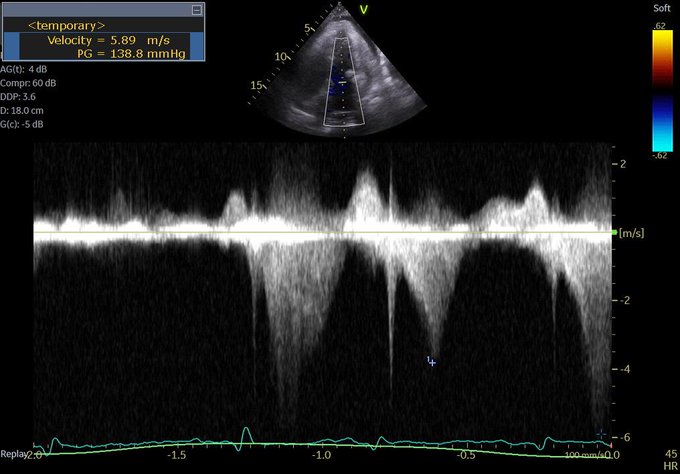
But first, let’s check out another CW tracing. Any thoughts?
This is a CW beam through LV apex and mitral valve – typically performed to assess mitral inflow and MR velocities and is part of the standard ECHO exam. However, the tracing is not typical for MR (late peaking, dagger shape). Remember, CW does not have depth resolution.
This is likely mid-cavitay/intra-ventricular obstruction. This is caused by complete mid-systolic obliteration of LV cavity (see PSAX) causing obstruction to the apical systolic flow. Again, seen in hypercontractile, underfilled, thick LV – e.g. sepsis (PMID: 26082197).
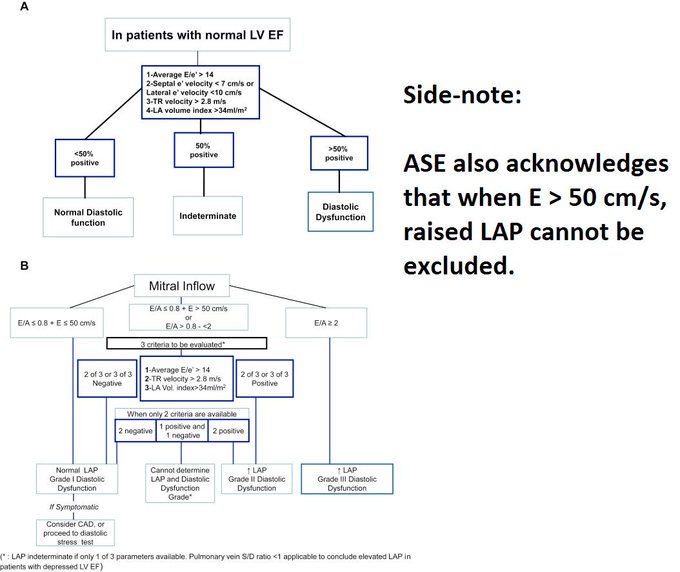
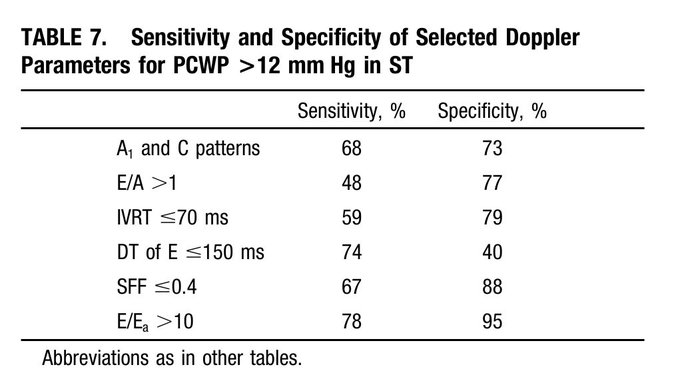
Finally – what does the ECHO tell us about LV filling pressures? – E/A ratio: As Lars pointed out, an E/A < 0.8 usually means normal LAP. However, the exception to this is sinus tach. This was shown in a study by none other than Dr. Nagueh (PMID: 9778330). (Also, see image)
The idea is that when early filling (E) is incomplete due to short diastolic time, the LA remains “full” at the time of the atrial kick – causing higher A velocities. NB: In that paper, E/E’ > 10 had a specificity of 95% for elevated LAP in ST. In our case: E/E’ = 75/5 = 15!
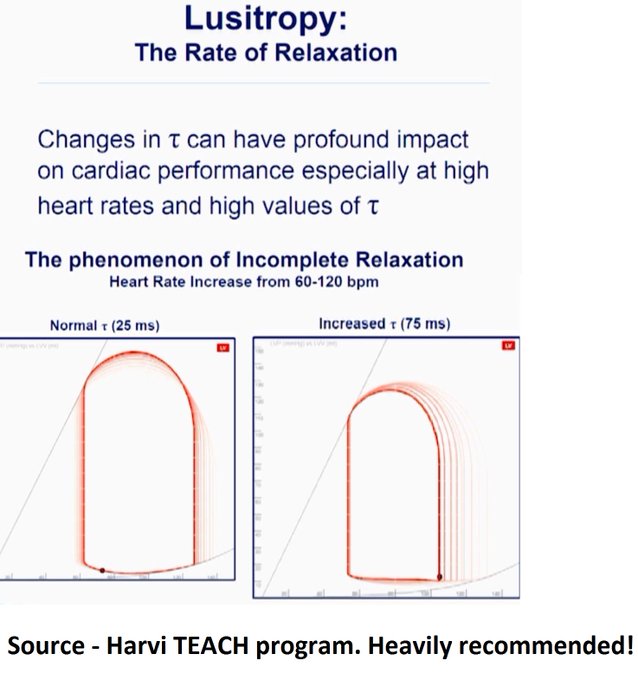
Potential contributors of high LAP – (i) SAM-associated MR – ‘trace’ in this ECHO but maybe we didn’t catch it. (ii) Tachycardia – E’ is 5 suggestive of delayed relaxation. Tachycardia causes “incomplete relaxation”. (iii) High afterload – high-grade dynamic obstructions.
So at this point, it’s still contentious but I have my money on hydrostatic pulmonary edema. Will detail our interventions and the remaining course in a bit. …Sorry to make this long but I think it’s worth it!
Now for the home stretch, the remaining course: We realized pericardiocentesis may be required soon but wanted to see if volume helps with (i) Peri-tamponade (ii) Dynamic obstructions. It helped a little – O2 requirements went from 60% HF to 6L NC. BP okay but still tachy.
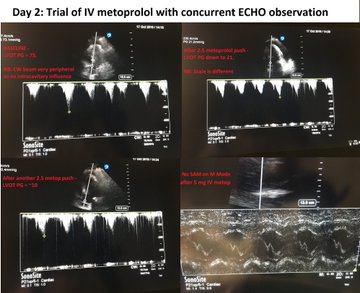
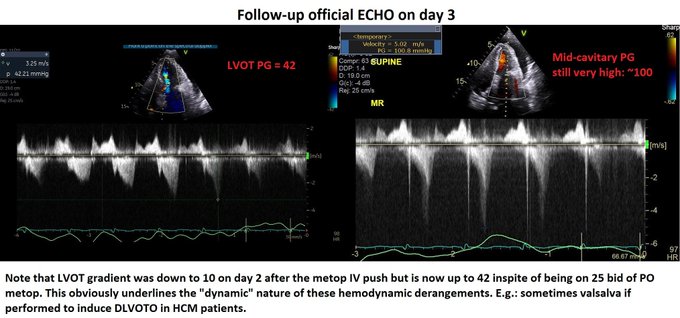
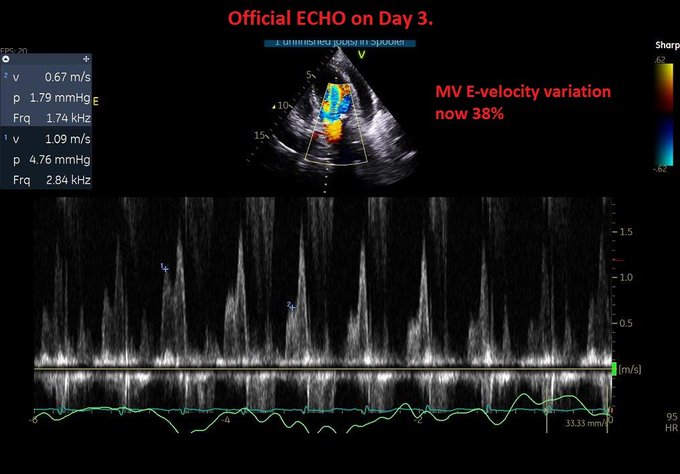
Day 2: We pushed 2.5 mg metop x2 with concurrent ECHO. LVOT gradient improved from 70s to ~10! (I did not compare mid-cavitary gradient, apologies). Started on 25 bid of PO metop later that night. HR now 90s Day 3: Official ECHO shows improved but persistent gradients.
Evaluation of tamponade was similar to previous ECHO but E-wave velocity variation now 38% –> elective pericardiocentesis: 550 cc removed. Fluid was transudate We also tapped a small pleural effusion pocket: transudate, cx negative (again goes with hydrostatic pulmonary edema).
Day 3 (contd): inc metop to 50 Q12H to blunt the gradients.
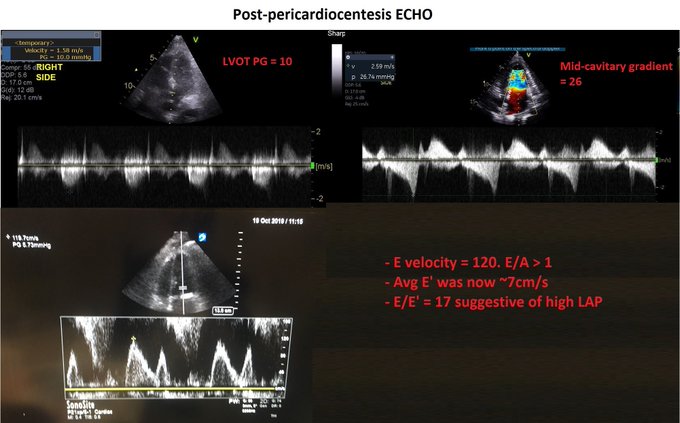
Day 4 – HR in 80s. ECHO shows no DLVOTO and non-significant mid-cavitary gradient. Oxygenation improved but still not normal. Why?! Check the E-velocity post-pericardiocentesis: it has jumped to 120 with E/A > 1.
So why is the LAP still high despite no significant dynamic obstruction? – Patients with chronic pericardial effusion may have chronically impaired diastolic filling –> low output –> volume retention (basic CHF physiology). When pericardial restraint suddenly released ––> increased LV preload –> high LAP.
Originally discussed elegantly here: PMID 6877287.
This is especially true if the LV has some baseline dysfunction. Day 5 – We started diuresis! The obvious risk was to precipitate the dynamic obstructions –> metop increased to 50 Q8H.
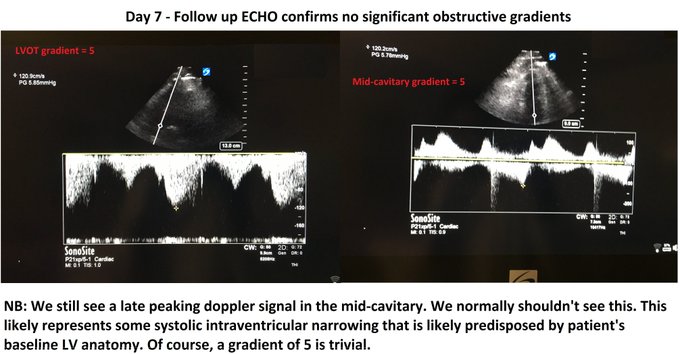
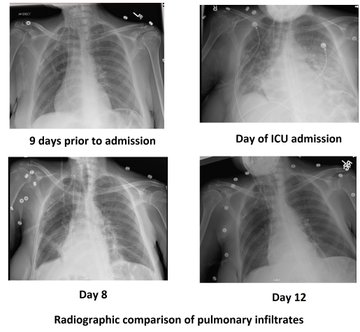
Day 7: Excellent diuresis (~2-3L negative per day). Hemodynamics stable (SvCO2 normal). Resting HR 60s – 70s. Follow-up ECHO confirmed no dynamic obstructions (see image). Day 8: Finally on room air. Pulmonary infiltrates improved (image). All cx remained negative.
Some dogmalysis offered by this case – – Fluids (probably) helped the pulmonary edema; CPAP/diuresis may have worsened. – IV metop contraindicated in hypotension? Not in this case – Sometimes you may have to diurese someone who recently had DLVOTO, as discussed above.
This case highlights the cognitive flexibility required to deal with hemodynamic puzzles. One thing I would’ve done different is be more aggressive with metop early on as it made a huge difference with DLVOTO. This was quite a ride. Hope you had fun. Feel free to share!
Much kudos to the treating team, I think this was excellently managed. As Amand says, cognitive flexibility ias absolutely key in assessing hemodynamics, particularly in the grey zones when multiple processes occur and co-exist. Managing this type of case using a recipe-based approach and without POCUS could have let to a poor outcome.
Now the POCUS used in this case is on another level. Very impressive and allowing incredible insight and certainly many potentially clinically useful Doppler analysis tips for LVOTO and LAP assessment.
In the end, I think that there were three pathologies, (a) tamponade physiology, (b) dynamic LVOTO, exacerbated by (c) hypovolemia (diarrhea) I might have approached this differently, had I seen a truly plethoric IVC. In such a case, one can easily see how tamponade physiology would contribute to LVOTO in two ways by creating intracardiac hypovolemia, hence worsening LVOTO both by decreasing LV preload and by the compensatory tachycardia. My first approach would probably have been to drain the pericardial effusion, and reassessing the hemodynamics afterwards, but correcting the intravascular deficit was necessary.
The other important thing this case re-emphasize is that tamponade is not a static diagnosis but a physiological spectrum. For the same given effusion (read intrapericardial pressure – IPP), it is the RA pressure that will determine whether overt tamponade develops. In this patient, it is very likely that a day earlier, there was no frank tamponade, but that after some diarrheal volume loss, the RAP dropped, and now IPP > RAP. It is important to know this because if you have an effusion and a fairly full IVC, one needs to be very careful with anything that can drop the RAP, meaning diuretics and vasodilators, because these can easily turn pre-tamponade into overt shock. And, as this case illustrates so well, you might even end up with LVOTO and pulmonary edema! Which is one of the myriad reasons one should have a basic POCUS exam in every acutely ill patient. These are things a resucitationist needs to know and prepare for.
So I recorded a chat with Domagoj (@domagojsono in the twitterverse), an anaasthetist-resuscitationist-intensivist from Freiburg a few months ago, but with H&R2019 and its aftermath, been slow in processing a lot of stuff I’ve got stocked… Apologies!
So in this one, DOmagoj and I discuss a bunch of resus topics, from eCPR to tissue oximetry. I’m really jealous of the fact that he does prehospital work with an ECMO van!!! …and with cool gear and of course, POCUS!
Here is the chat, hope it leads to thoughts, discussion and contribution!
So last week sometime we had an interesting twitter exchange which made me realize it is important to explain how some of us are using venous POCUS in different clinical scenarios, which is key, because the development of monosynaptic clinical reflexes with POCUS findings is a rabbit hole we should try not to go down. Instead, POCUS should be about asking the right question and taking that answer as a piece of the pathophysiologic puzzle facing us, which may mean intervening sometimes, and sometimes not, for the same given finding, but with different surroundings.
Thanks to those involved in that discussion – it is how we grow!
And here are some thoughts:
For those not up to speed on venous congestion POCUS I put up the chapter that Korbin Haycock, Rory Spiegel and I worked on in this earlier post.
Here are Korbin’s thoughts on this:
I’m very glad Dr. Eduardo Argaiz pointed this case out, as it brings up considerations apropos both chronic venous congestive cases as well as management of acute illness, particularly in sepsis, where we would expect patients to most likely be fluid responsive, but fluid tolerance is largely overlooked with current management strategies by the majority of clinicians.
Phil’s above audio commentary points out the difference is these two broad categories very nicely. If you didn’t listen to it–you should.
With respect to chronic venous congestive conditions, the knowledge and application of Doppler assessment to therapy will hopefully be the next advance in management at large. Already, I think there is more than adequate research available to show the value of Doppler POCUS (D’POCUS, D/POCUS, or DPOCUS?) in managing these patients. It’s only a matter of clinicians willing to commit to learning and integrate this technology into their skill set.
With respect to resuscitation of the acutely ill patient, there is by far less data, and we are probably into the realm of N=1 here, in terms of how to manage these patients. But, I personally believe–and I understand this is my opinion–that current trends in resuscitation (especially sepsis resuscitation), largely ignores the effect of over volume resuscitation and the potential downstream damage inflicted on our patients.
This theoretical damage of over aggressive fluid resuscitation is multifactorial, including glycocalyx shedding issues/endothelial dysfunction, positive fluid balance and EVLW causing increased mortality (which there is ample evidence for, I think), venous congestion leading to perfusion injuries to encapsulated organs, such as the kidney (AKI) and brain (congestive encephalopathy), and end organ edema leading to the perpetuation of a malignant inflammatory syndrome (portal HTN and gut edema).
In the case called out by Dr. Argaiz, (which can be reviewed by the previous post on this website) my patient had an IVC that whilst not plethoric, was not an IVC that one would expect to find in a patient with a typical distributive shock pattern (i.e. increased cardiac output, decreased SVR, and decreased RAP). Firstly, the complicating factor of atrial fibrillation with RVR was central to the patient’s shock state, however this was quickly addressed with rate control. However, in addition, this particular patient did exhibit additional signs of venous congestion. The portal vein was pulsatile and the intrarenal Doppler pattern was interrupted/bi-phasic in nature. Granted, a pulsatile PV Doppler could be interpreted as related to the hyper dynamic nature of septic shock (as the esteemed Dr. Denault correctly cautioned in his comments on the original post), however a less than flat IVC and the intrarenal findings gave weight to a venous congestive hypothesis as a cause the PV findings as well as a possible cause for his AKI evident on his initial labs.
With this particular case, given my personal global POCUS/FOCUS assessment of his increased LAP (high E/e’), RV dysfunction, RAP, PV, and intrarenal Doppler venous pattern, AND that fact that the RRI was insanely high with an AKI, I elected to treat my hypothetical construct of his renosarca with furosamide and his RRI with vasopressin (as the NE infusion did increase his MAP, BUT NOT decrease his RRI–which the vasopressin infusion did decrease, or so I presume as no other therapeutic interventions were given with respect to the time frame the RRI decreased).
In the end his kidneys had recovered by the next morning, which I’m sure that any intensivist will admit is the opposite of the norm, as the kidneys usually get, at least transiently worse initially-being the delicate sissies/whimps that they are. Whether this was because of the diuretic or the vasopressin, or something else, is debatable for sure, but it sure didn’t get better by 30 cc/kg of crystalloid mandated by CMS, because he got not a drop more than what was needed to push the diltiazem, the lasix, the antibiotics, and the vasopressors.
So to summarize, in the case of chronic cardiogenic venous congestion, clinician realization and adoption of Doppler assessment of this entity will likely be the next leap in improvement in the management of these patients. In the case of acute resuscitation, venous congestion may be a bit more nuanced, and a more comprehensive evaluation is in order in a case by case fashion. However, I think recognition of the issues of over aggressive volume administration will probably be the next frontier in sepsis resuscitation.
So given the importance of these topics, the number of questions and discussions we’ve had on the twitterverse, and most importantly in the spirit of #FOAMed, here is the chapter from the POCUS book which was co-authored by Rory Spiegel (@EMnerd), Korbin Haycock (@korbinhaycockmd) and myself.
So I keep hearing and seeing people bash the IVC. Casually dismissing it with a shrug. “It’s not really good for volume responsiveness, you know…”
All that deserves is an eyeball-rolling emoji. That is, unfortunately, the reaction of docs who are trying to devise a threshold or recipe-based approach to POCUS management (which will be just as bad as any recipe-based medicine) as opposed to physiological understanding of what is going on with the patient.
There’s so much good information packed in scanning the IVC (properly, in both axes – for more, see a bunch of my previous posts), and frankly, volume responsiveness is the least of my concerns, that it is a shame to toss out the proverbial baby with the bathwater.
So I talked about this at Stowe EM – an awesome conference run by my friend Peter Weimersheimer (@VTEMsono), which I highly recommend to anyone for next year, great talks, people and spot:
Oh yes, and anyone looking to explore physiological, evidence-based, cutting- and bleeding-edge approaches to resus, don’t miss H&R2019 this May in Montreal!
Definitely interesting stuff, and have to commend the authors on a complex resuscitation strategy that had some real-world flexibility built in in terms of later generalizability and applicability for real-world cases. However there are some fundamentals I have concerns about. Let’s see what Rory thinks:
Yeah. I think the bottom line of opening resuscitationists’ eyes to NOT apply monosynaptic reflexes of giving fluids to elevated lactate is good. In that sense, definitely a step forward.
However, the insistence on maximizing CO under the illusion of optimizing perfusion remains problematic and leads to a congested state unless only a small or perhaps moderate amount of fluid is required to achieve non-volume responsiveness. I think it’s important to realize that the most rapid correction of hemodynamics is a surrogate marker and has not been definitively associated with survival across the board (eg the FEAST study and others), and it’s only proven clinical impact may be on health care workers’ level of anxiety.
Tune in soon for some other smart docs’ take on this!
So, fresh from reading Jon’s post, I felt I had to add a bit of nuance in my previous post to what I feared some might extract as a take-home message, even if in fact, we are not that differing in opinion at all – which Jon expressed here:
i agree with ultrasound for finding the uncommon causes of shock. these examples seems to permeate twitter and make ultrasound very appealing. because ultrasound is non-invasive, it makes the risk-to-benefit ratio very low for these uncommon but highly-lethal and treatable causes.
but that needs to be compared to the risk-to-benefit ratio of ultrasound for the more common causes of shock – like ‘non-cardiogenic, septic’ etiologies as seen in SHOC-ED. here, “static’ ultrasound [as per the RUSH and ACES protocols] – per SHOC-ED – appears to be neither helpful nor harmful. your read of the discussion is perfect, but i was depressed because it read as if the authors only realized this ex post facto – study of previous monitoring utensils [e.g. PAC] should have pre-warned the authors …
i will take some mild issue with markers of volume responsiveness and tolerance. you are correct on both fronts – but what the data for the IVC reveals – perhaps paradoxically – is that true fluid responders can have a very wide-range of IVC sizes from small to large and unvarying … this was born out in most of the spontaneously breathing IVC papers [airpetian and more recent corl paper] the sensitivity was rather poor.
the same *could* be true for the opposite side of the coin. a large great vein may not mean a volume intolerant patient. i tried to exemplify how that could be so in the illustrative case in my post. an elderly man, with probable pulmonary hypertension and chronic TR who probably “lives” at high right-sided pressures. nevertheless, he likely has recurrent C. diff and is presenting 1. hypovolemic and 2. fluid responsive despite his high right-sided pressures. portal vein pulsatility *could* be quite high in this patient – but he still needed some volume.
the obvious underlying issue here – which I know you are well attuned to – is that a Bayesian approach is imperative. when you PoCUS your patients, you are inherently taking this into consideration – i know that you are a sophisticated sonographer. my hidden thesis of the post is that if ultrasound findings are followed in a clinical vacuum and followed without really understanding the physiology [which can explain clinico-sonographic dissociation – like the patient in my fictitious case]… disappointment awaits.
Then Korbin Haycock chimes in and adds a level of understanding that I completely agree with but had difficulty in expressing, but which I think is key to understanding the current and future evolution of POCUS. Complex, operator-dependant medical leaps such as laparoscopic surgery suffered with similar growing pains. But I’ll let Korbin shed some light:
I think the issue of POCUS in resuscitation is somewhat analogous to Prometheus’s gift of fire to humanity.
Jon has quite aptly pointed out that if POCUS (particularly a single POCUS supplied data point such as IVC diameter), if used in isolation, without clinical context, and without comprehensive information, is not much better than using a single data point such as CVP to make complex clinical decisions. Multiple factors influence the behavior of the IVC, just as they do with the CVP. Being a dynamic entity, the IVC does have some advantages over a static number like the CVP. However, if considered by itself, the IVC POCUS evaluation will only result in the same pitfalls as using the CVP as a guide to fluid management. If POCUS is applied in such a blunt manner, we are doomed to repeat our previous folly of using the CVP as a guide to fluid resuscitation. I hope I am in the ball park of the core of Jon’s point here, if not as very eloquently stated by him.
Phil is advocating a more nuanced and sophisticated approach to POCUS than what the SHOC-ED trial investigators used to guide management in their study. Most shocked patients presenting to the ED (“Emerge!”) come with a phenotype of distributive shock. Indeed, these were the majority of the patients in the SHOC-ED trial. Any experienced clinician will recognize this syndrome virtually every time, with no more than an “eyeball and Gestalt” assessment from across the room and a set of vital signs. Current dogma is that this syndrome ought to be treated with 30 cc/kg of crystalloids and then to add a vasopressor if the patient’s blood pressure is still low. Given this, there couldn’t have been much difference as to how patients were managed in either group in this study. I however, disagree with this aggressive crystalloid administration approach, as I’m sure many readers of Phil’s blog do as well. What I gather Phil is saying here is, as he insightfully stated in the past, “IVC never lies, it’s just not telling you the whole story.” A complete POCUS gives us (OK, well almost) the whole story. The caveat here is you must know a whole lot about POCUS. Thus the Prometheus analogy. A little information is a child playing with fire.
Someone new to POCUS, with only a novice’s understanding of what an IVC POCUS evaluation means, will probably make the correct assessment of a patient’s fluid status about 60-70% of the time. This probably is only slightly better than an experienced clinician’s non-POCUS judgement. Hardly enough to translate into any meaningful clinical outcome in a trial without a ridiculously large sample size to find a pretty small benefit. But POCUS potentially offers so much more information. LV and RV systolic function, LV and RV diastolic function, SV, CO, SVR, PVR, RAP/CVP, sPAP/mPAP/dPAP, LVEDP/LAP/PAOP, valvular pathology, tamponade, fluid responsiveness (for what ever that’s worth!), RV/LV interactions (both in series and in parallel), EVLW, insight into pulmonary vascular permeability, renal resistive index/renal venous congestion, portal hypertension/congestion, gut flow resistance, and on and on. Most of this information can be more or less determined in less time that it takes to put in a central line in order to get the damned CVP (actually, I do like to know what my CVP is, for what it’s worth). The more data points you are able to collect with increased POCUS skills and experience, the more grasp you have as to what is going on with your patient and the right way to treat them. I would argue that given the information attainable with advanced POCUS skills, POCUS is a no-brainer that will enormously improve not only individual patient outcomes, but effect populations at large, if only the general hospital based practitioner can attain a more than introductory understanding of POCUS.
So, I guess the question is, “how much training is enough training?” I don’t know. Inevitably, POCUS knowledge will incur a bit of the Dunning-Kruger effect as pointed out by Jon’s example of an IVC POCUS fail. But reading Jon’s clinical case example, from the get go, I found myself asking questions that would change may management one way or another with additional information that I could get quickly and easily with additional POCUS interrogation of the patient. Jon pointed this out himself by revealing that the patient has pulmonary hypertension as manifested by the tricuspid regurgitation upon auscultation of the heart. With POCUS, I don’t need to guess what a heart murmur is or how bad it is or even if it is relevant to my patient in this case for that matter. POCUS can tell me it’s TR and it tells me what the sPAP/mPAP/dPAP and PVR is if I care to find out. So if this level of information can be gleaned, for me, no one can argue that POCUS has no merit. But, I’ve spent a lot of time striving to be good at this, just as probably a lot of people reading this have done as well. What about newbies?
Consider: At my main hospital, for a variety of sensible reasons I won’t get into, we decided to train a group of nurses in POCUS in order to evaluate septic patients. They achieve basic training in POCUS and are very competent sonographers with regard to IVC, gross LV and RV function, and pulmonary edema. They are a small group of very intelligent, skillful nurses that are excited to learn all they can. We had them evaluate every septic patient that presented to our hospital, do a POCUS exam, and discuss the findings with a physician. We established some very basic resuscitation endpoints largely based on POCUS findings applied to each individual patient and their POCUS exam. Our severe sepsis/septic shock mortality rates dropped from 35-38% to 8-10% with this program. Our hospital plans to publish this data officially soon for public analysis, but it did make a difference in our experience. That said, my nurses do frequently show me cases where I notice some small detail on their POCUS exam that propmts an additional investigation that alters the plan in management. Also, some of my very competent POCUS savvy residents make errors because they don’t have enough knowledge yet. I’m sure I can make these errors too at times as well, although hopefully less and less so with time.
Here’s my point: Heed Jon’s admonition to look at the big picture and not rely on isolated data points. Be inspired by Phil’s passion for the potential of a good POCUS evaluation. If you only get your toes wet with POCUS, you are playing with forbidden fire. But if you care to look into it further, POCUS opens up worlds to you. By all means, learn all you can about POCUS. Recognize that if you are new to POCUS techniques, there are improtant caveats to each finding, and physiology that needs to be considered with a comprehensive view, some of it may be strictly non-POCUS related information as well. Your patient is unique and only a careful comprehensive consideration of what’s going on with your patient will guide the best approach to your management of their illness. I don’t think SHOC-ED or any other trial for that matter can address the nuances of good individualized patient management. That is up to you.Jon replies:
nice analogy – i think Korbin’s response is appropriate and i look forward to speaking alongside him in May. as i chew on the SHOC-ED a little and try to distill my concerns – i think what it boils down to is this: it’s less about playing with fire – i think – and more about how this fire is brought to the community as a whole. my post on pulmccm was more of a warning to the early adopters [like us] who are planning these trials. imagine 40 years ago:
-the flotation PAC is introduced, a small group of clinical physiologists use it thoughtfully, understand the caveats, the problems of data acquisition, interpretation, implementation, the problems with heart-lung interactions, intra-thoracic pressure, etc.
-these early adopters present their results to the community as a whole
-the physiology of the PAC is simplified
-the numbers from the PAC are introduced into algorithms and protocols and **widely** adopted into clinical practice
-the PAC is studied based on the above and found to make no difference in patient outcome.
-in 2010 a venerable intensivist suggests floating a PAC in a complicated patient and the fellow on rounds chuckles and states that their is ‘no evidence of benefit’
does this sound eerily familiar? is our present rhyming with the past? if the planners of POCUS trials are not careful, i promise you that the same will happen but insert any monitoring tool into the place of PAC. i can very easily visualize a fellow on rounds in the year 2030 scoffing at the idea of PoCUS because trials [SHOC-ED, and future trials x, y and z] showed no difference in patient outcome. is it because PoCUS is unhelpful or is it because the way it was introduced and studied was unhelpful? and the three of us will sound like the defenders of the PAC from 30 years ago: “PoCUS isn’t being used correctly, it’s over-simplified, it works in my hands, etc. etc.”
it’s not PoCUS that’s unhelpful, it’s how we’re implementing it – and i was most depressed when the authors of SHOC-ED appeared to stumble upon this only in the discussion of their paper – like you mentioned phil. imprecise protocols will result in equally imprecise data and the result will be nebulous trial outcomes. we should all be worried.
Korbin adds:
Excellent points Jon. The PAC example is very relevant, as on more than one occasion, I’ve had the argument put to me by some colleagues that essentially how I’m applying POCUS is really no different than the information gleaned from the PAC, and “that’s been shown to not be helpful to outcomes” etc. So, therefore, why do I bother?
Then again, I’ve seen a fair amount of phenylephrine being thrown at hypotensive cardiogenic shock patients after a 2 liter normal saline bolus didn’t do the trick.
You are absolutely spot on when you point out that seeing the big picture, knowing the physiology, and being aware of the pitfalls of isolated data points is important to making the right decisions in patient care.
Furthermore, I agree that when a clinical trial is done that doesn’t consider some of the nuances of all this, and “shows” that POCUS, or any other diagnostic modality for that matter, doesn’t contribute to better patient outcomes, it probably only serves to marginalize a potentially valuable diagnostic tool to an actually astute intelligent clinician.
I’m not meaning by saying this to bash the good intentions of the SHOC-ED trial. To be fair, it’s really hard to design a trial that can take into account all the permutations that are involved in any individual patient presents with, having their own unique clinical situations, hemodynamic profiles, co-morbidities (both known and undiagnosed), etc. POCUS, PAC, transpulmonary thermodilution, ECG, chest x-ray, CT scans, labs, physical exam–these are all merely tools that guide patient care. Albeit some are way more powerful than others. I can image various amounts of uproar if some of these traditional tools were subjected to clinical trials to prove their utility. The argument, if proven “useless” in a study for the oldest and well accepted tools would always be, “put it in the clinical context, and its value speaks for itself.” For me, I’d happily like to make clinical descisions based on information based on an advanced POCUS exam or PAC, rather than interpreting hepatojugular reflux or a supine chest x-ray.
Any diagnostic test requires that the clinician understand the limitations of that test, and understand that the whole clinical scenario must me taken into account. You’ve hit on that, I think, with your argument. This surely has implications when any technology or test is studied.
‘Nuff said.
Philippe
PS These are just the kind of discussions that can change both the way you approach medicine and manage your patients, and these are the ones you find behind the scenes and in the hallways of H&R2018. Don’t miss H&R2019 if you take care of sick patients. It’s the kind of small, chill conference where the faculty will be happy to take a few minutes and discuss cases and answer all your questions (if they can) about acute care.
Registration is open and we have said goodbye to the snail mail process. Fortunately, we are a lot more cutting edge in medicine than in non-medical technology.
We are really excited about this programme, and a lot of it comes from the energy and passion coming from the faculty, who are all really passionate about every topic we have come up with.
The hidden gem in this conference is the 4 x 40 minutes of meet the faculty time that is open to all. Personally I’ve always felt that I learn so much from the 5 minute discussions with these really awesome thinkers and innovators, so wanted to make it a priority that every participant should get to come up to someone and say ‘hey, I had this case, what would you have done?’ Don’t miss it!
CME Accreditation for 14 hours of Category 1.
This programme has benefitted from an unrestricted educational grant from the following sponsors (listed alphabetically):

















So, fresh from reading Jon’s post, I felt I had to add a bit of nuance in my previous post to what I feared some might extract as a take-home message, even if in fact, we are not that differing in opinion at all – which Jon expressed here:
i agree with ultrasound for finding the uncommon causes of shock. these examples seems to permeate twitter and make ultrasound very appealing. because ultrasound is non-invasive, it makes the risk-to-benefit ratio very low for these uncommon but highly-lethal and treatable causes.
but that needs to be compared to the risk-to-benefit ratio of ultrasound for the more common causes of shock – like ‘non-cardiogenic, septic’ etiologies as seen in SHOC-ED. here, “static’ ultrasound [as per the RUSH and ACES protocols] – per SHOC-ED – appears to be neither helpful nor harmful. your read of the discussion is perfect, but i was depressed because it read as if the authors only realized this ex post facto – study of previous monitoring utensils [e.g. PAC] should have pre-warned the authors …
i will take some mild issue with markers of volume responsiveness and tolerance. you are correct on both fronts – but what the data for the IVC reveals – perhaps paradoxically – is that true fluid responders can have a very wide-range of IVC sizes from small to large and unvarying … this was born out in most of the spontaneously breathing IVC papers [airpetian and more recent corl paper] the sensitivity was rather poor.
the same *could* be true for the opposite side of the coin. a large great vein may not mean a volume intolerant patient. i tried to exemplify how that could be so in the illustrative case in my post. an elderly man, with probable pulmonary hypertension and chronic TR who probably “lives” at high right-sided pressures. nevertheless, he likely has recurrent C. diff and is presenting 1. hypovolemic and 2. fluid responsive despite his high right-sided pressures. portal vein pulsatility *could* be quite high in this patient – but he still needed some volume.
the obvious underlying issue here – which I know you are well attuned to – is that a Bayesian approach is imperative. when you PoCUS your patients, you are inherently taking this into consideration – i know that you are a sophisticated sonographer. my hidden thesis of the post is that if ultrasound findings are followed in a clinical vacuum and followed without really understanding the physiology [which can explain clinico-sonographic dissociation – like the patient in my fictitious case]… disappointment awaits.