
So #MedTwitter is truly an incredible forum for case discussion, where you get to exchange with literally some of the best medical minds on the planet who often also happen to be front-line clinicians in the nitty-gritty therapeutic decision-making. Here’s a discussion which I think was great. Recently, Dr. Thind has been generating some great cases and hemodynamic discussions. I thought this one was worth highlighting!

Dr Thind is an internist and currently Critical Care Hospitalist (and upcoming ICU fellow) at the Cleveland Clinic, and tweets out some great #FOAM from @Thind888 on twitter.
Case:
OK, let’s give this a shot. Here’s a ‘hemodynamics special’. Saw this case a couple weeks ago. A lot of decision making was based on educated guesses so it should be a good one for discussion. – 51 yo woman being worked up on the floor for chronic diarrhea, moved to ICU for hypoxia.
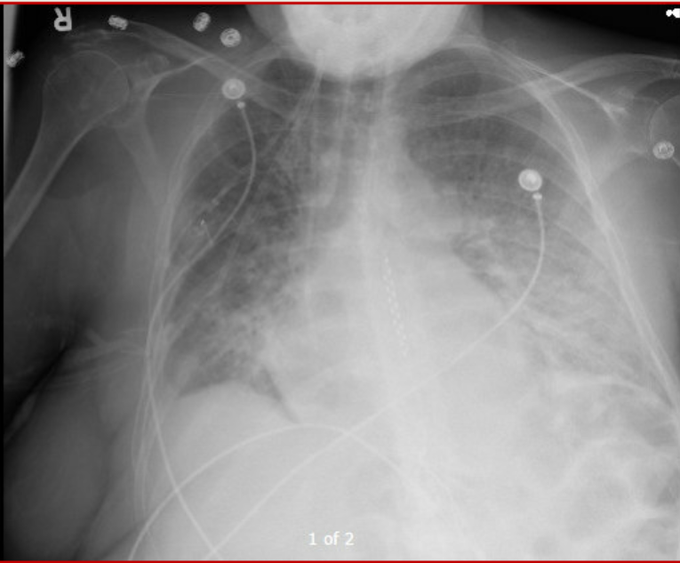
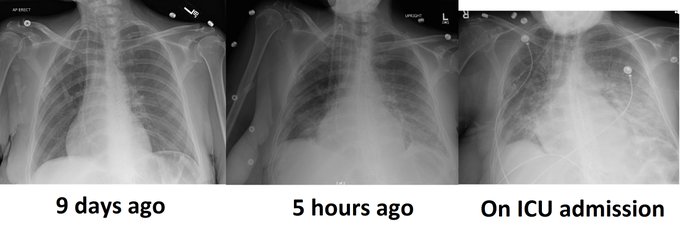
Dyspnea progressed over few hours. Vitals significant for tachycardia (140s) and hypotension (MAP in low 60s). On arrival, SBP 60s – improved with fluid bolus. CXR attached. Patient has H/O of pericardial effusion for several months that has been managed conservatively.
The patient has an official ECHO performed on arrival in ICU (images attached). IVC difficult to assess but about 2cm without collapse. Lung US – diffuse B lines.
OK so right there a flag goes up for me. A plethoric IVC means something is wrong. Sounds too vague maybe, but you need to find the reason for this, as it likely has therapeutic implications. Let’s see what comes up.
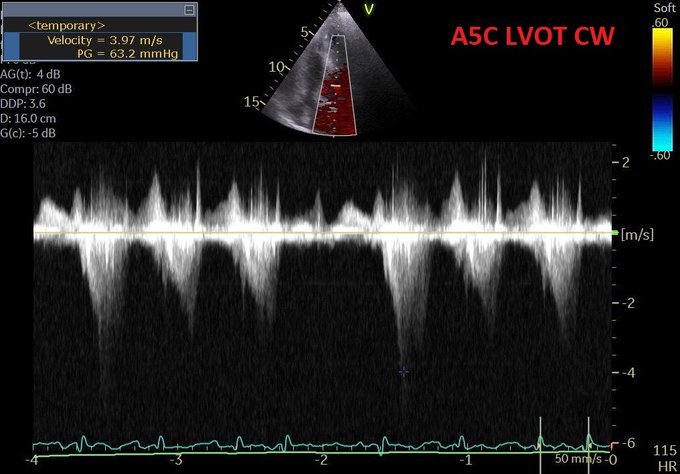
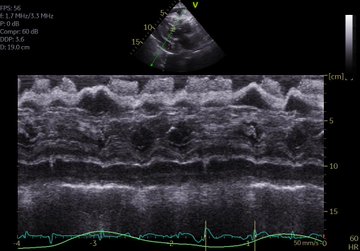
Modifed A5C.
LVOT doppler

CXR
Pressing questions –
(i) Is it hydrostatic or increased permeability pulmonary edema?
(ii) Fluids, diuresis, or none?
(iii) Would CPAP help?
(iv) Drain the pericardial effusion?
(v) What about that LVOT doppler?
Mitral inflow velocities and TDI attached. M-mode through PLAX almost uninterpretable. Lung infiltrates are new so less likely lymphangitic carninomatosis. Note: ScVo2 = 40s. Another Q to ponder on –
(vi) Is tamponade typically associated with hydrostatic pulmonary edema?
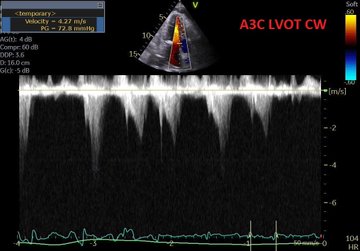
Perhaps this slowed up (0.5x) A3C loop will help with that LVOT doppler!
Great discussion as expected. Lets discuss:
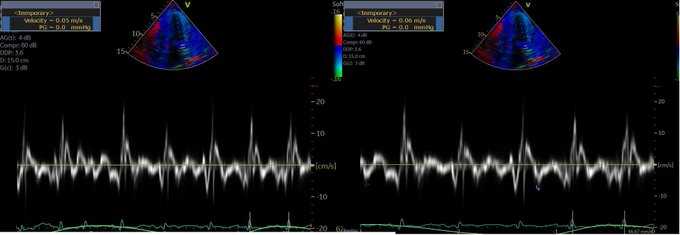
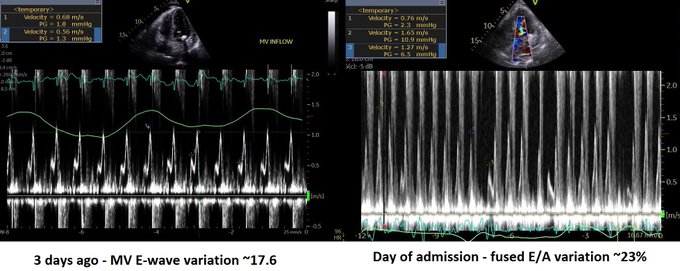
Q4. Is it tamponade? – This is not a slam dunk. Chamber collapse can sometimes be controversial. In these situations I try my best to get MV E-wave variation. I think our tech got a decent signal. But note these are fused E/A waves.
The first thing I look at to screen for tamponade is the IVC. Tamponade is an obstructive form of shock, dependant on the intrapericardial pressure exceeding the right atrial pressure. If it does, unless respiratory efforts are extreme, the IVC should become plethoric. Hence, the absence of such would make the effusion – given the current RA pressure – NOT tamponade. Yet again, another point scored by the IVC for usefulness.
Although I don’t see why we can’t use fused waves for this purpose (couldn’t find anything on it in the literature). Note that in spite of the cardiac motion, the mitral inflow variation is <25% (~23%). It’s close though, and certainly seems to have increased from 3 days ago.
The cardiologist (understandably) was non-committal and read it as “possible early tamponade”.
Q5. What about LVOT doppler? A good M-mode could not be obtained but the A3C in 6/ shows SAM. The report mentioned “chordal SAM” but I think you can clearly see “valvular SAM” too.
Chordal SAM is SAM of the chordal apparatus (you could see it bumping against the septum in 6/). It is (typically) NOT hemodynamically significant (PMID: 27241937). – When we see mitral SAM, it is important to quantify its hemodynamic effects – with LVOT peak gradient via CW.
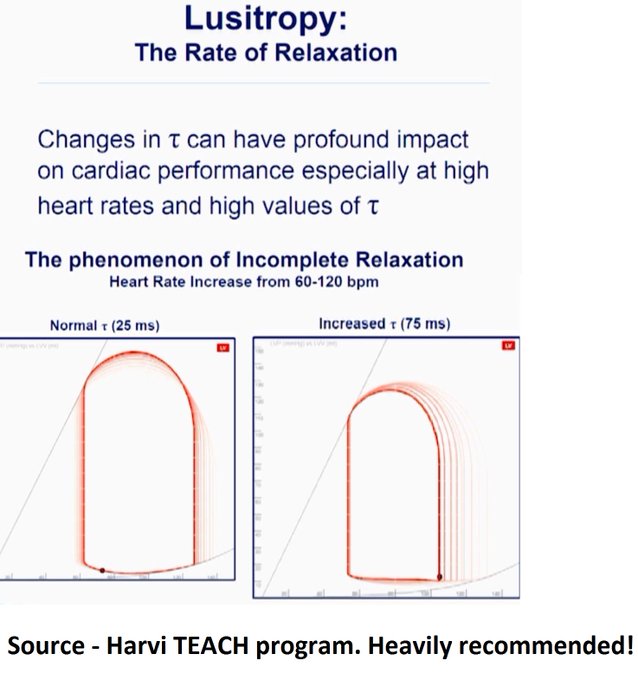
In HOCM, DLVOTO is defined by an LVOT gradient of >30; >50 is considered severe. Our patient had a gradient of ~70. Although classically a/w HCM, SAM can be seen in anyone with thick, hypercontractile, underfilled LV. Tachycardia further hampers LV filling (PMID: 27726435).
Mitral SAM is often a/w MR – this acute MR can cause flash pulmonary edema. These patients may actually need fluids (to help with SAM) to fix there hydrostatic pulmonary edema!! (PMID: 20661209). However, our patient only had trace MR (you could see it in 1-2 CD frames).
Working theory (similar to Lars) – Chronic stable pericardial effusion –> diarrhea (pt had 15 BMs the day before the admission) –> reduced venous return –> brought the patient at the verge of low-pressure tamponade (PMID: 16923755) –> further reduction in LV filling —> reduced stroke volume –> adrenergic drive causing tachycardia and increased inotropy –> all factors culminating in mitral SAM and DLVOTO.
This also explains the low ScVO2. Note – CPAP would further reduce venous return (Q3) so wouldn’t help, may hurt.
Now the most important Qs: why pulmonary edema and what to do about it (Q1 and 2). As tamponade causes impedance to venous return, it is not typically associated with high LAP and hydrostatic pulmonary edema (Q6).
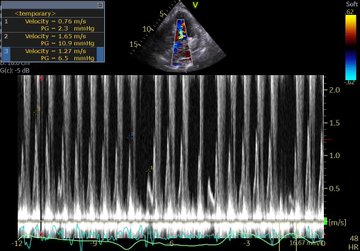
But first, let’s check out another CW tracing. Any thoughts?
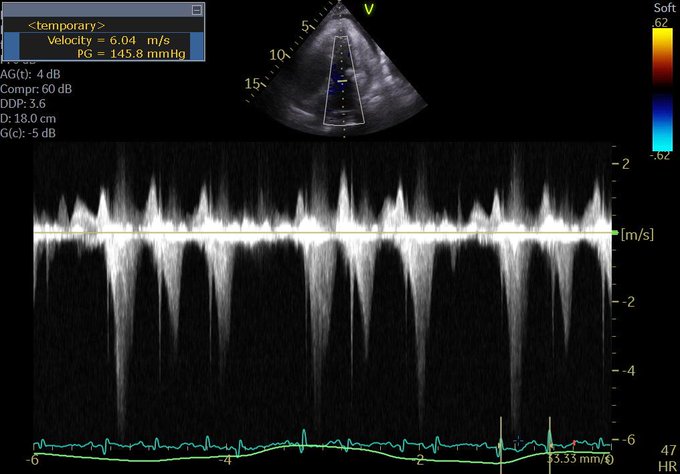
This is a CW beam through LV apex and mitral valve – typically performed to assess mitral inflow and MR velocities and is part of the standard ECHO exam. However, the tracing is not typical for MR (late peaking, dagger shape). Remember, CW does not have depth resolution.
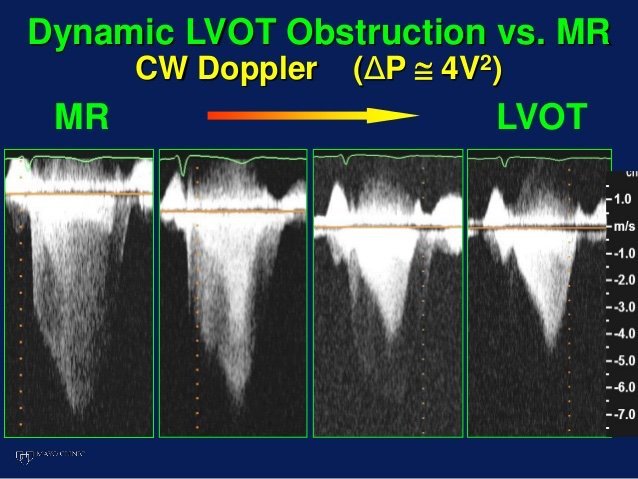
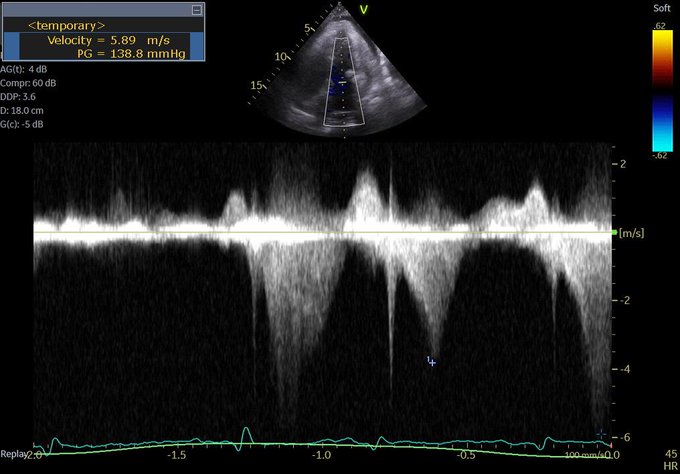
This is likely mid-cavitay/intra-ventricular obstruction. This is caused by complete mid-systolic obliteration of LV cavity (see PSAX) causing obstruction to the apical systolic flow. Again, seen in hypercontractile, underfilled, thick LV – e.g. sepsis (PMID: 26082197).
Finally – what does the ECHO tell us about LV filling pressures? – E/A ratio: As Lars pointed out, an E/A < 0.8 usually means normal LAP. However, the exception to this is sinus tach. This was shown in a study by none other than Dr. Nagueh (PMID: 9778330). (Also, see image)
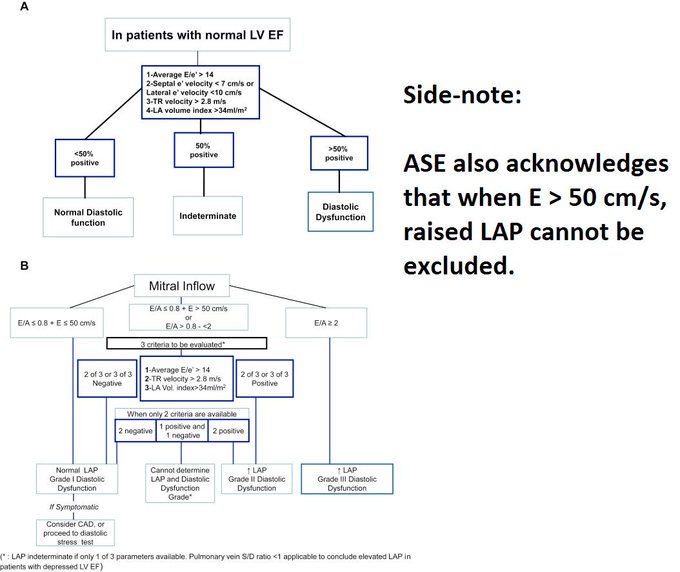
The idea is that when early filling (E) is incomplete due to short diastolic time, the LA remains “full” at the time of the atrial kick – causing higher A velocities. NB: In that paper, E/E’ > 10 had a specificity of 95% for elevated LAP in ST. In our case: E/E’ = 75/5 = 15!
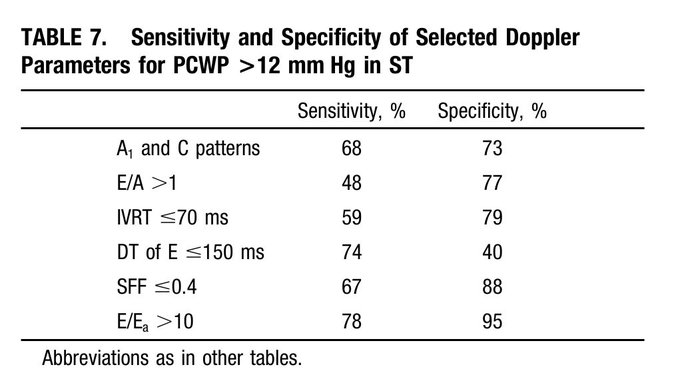
Potential contributors of high LAP – (i) SAM-associated MR – ‘trace’ in this ECHO but maybe we didn’t catch it. (ii) Tachycardia – E’ is 5 suggestive of delayed relaxation. Tachycardia causes “incomplete relaxation”. (iii) High afterload – high-grade dynamic obstructions.
So at this point, it’s still contentious but I have my money on hydrostatic pulmonary edema. Will detail our interventions and the remaining course in a bit. …Sorry to make this long but I think it’s worth it!
Now for the home stretch, the remaining course: We realized pericardiocentesis may be required soon but wanted to see if volume helps with (i) Peri-tamponade (ii) Dynamic obstructions. It helped a little – O2 requirements went from 60% HF to 6L NC. BP okay but still tachy.
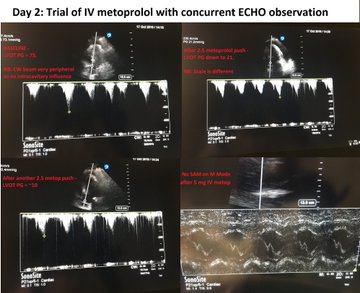
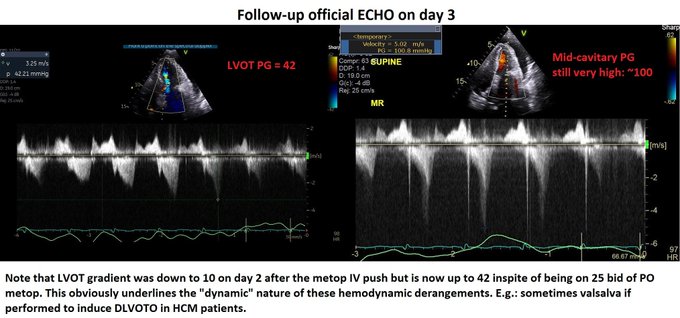
Day 2: We pushed 2.5 mg metop x2 with concurrent ECHO. LVOT gradient improved from 70s to ~10! (I did not compare mid-cavitary gradient, apologies). Started on 25 bid of PO metop later that night. HR now 90s Day 3: Official ECHO shows improved but persistent gradients.
Evaluation of tamponade was similar to previous ECHO but E-wave velocity variation now 38% –> elective pericardiocentesis: 550 cc removed. Fluid was transudate We also tapped a small pleural effusion pocket: transudate, cx negative (again goes with hydrostatic pulmonary edema).
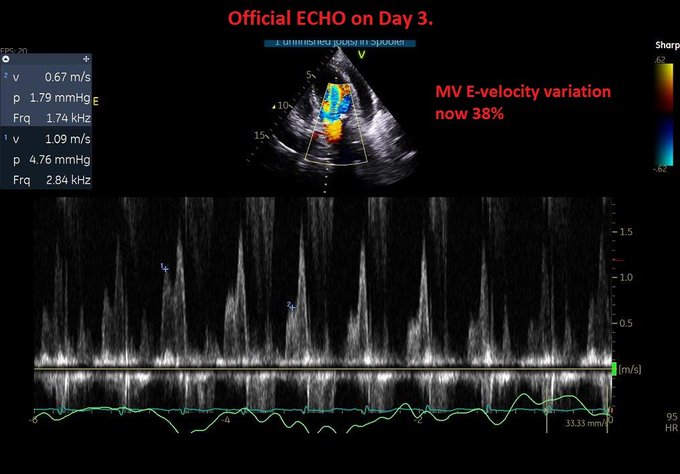
Day 3 (contd): inc metop to 50 Q12H to blunt the gradients.
Day 4 – HR in 80s. ECHO shows no DLVOTO and non-significant mid-cavitary gradient. Oxygenation improved but still not normal. Why?! Check the E-velocity post-pericardiocentesis: it has jumped to 120 with E/A > 1.
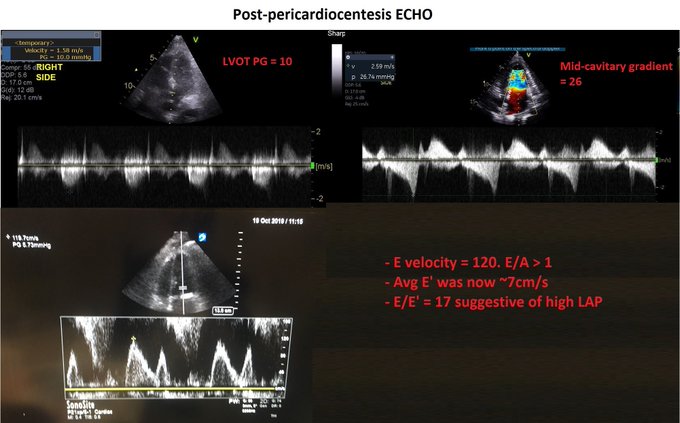
So why is the LAP still high despite no significant dynamic obstruction? – Patients with chronic pericardial effusion may have chronically impaired diastolic filling –> low output –> volume retention (basic CHF physiology). When pericardial restraint suddenly released ––> increased LV preload –> high LAP.
Originally discussed elegantly here: PMID 6877287.
This is especially true if the LV has some baseline dysfunction. Day 5 – We started diuresis! The obvious risk was to precipitate the dynamic obstructions –> metop increased to 50 Q8H.
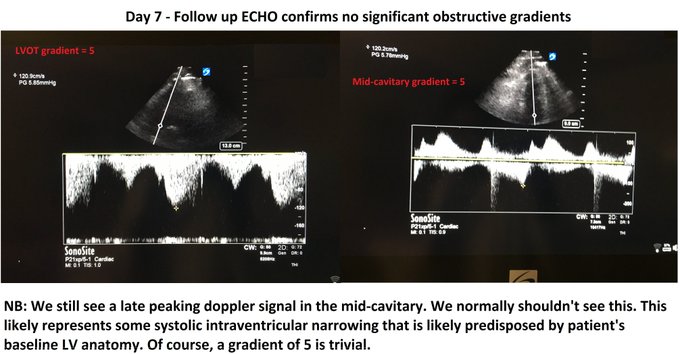
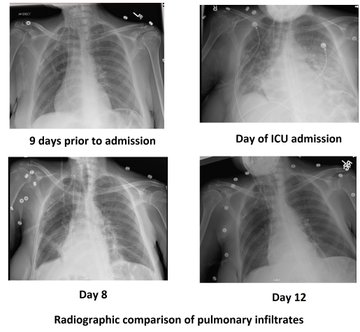
Day 7: Excellent diuresis (~2-3L negative per day). Hemodynamics stable (SvCO2 normal). Resting HR 60s – 70s. Follow-up ECHO confirmed no dynamic obstructions (see image). Day 8: Finally on room air. Pulmonary infiltrates improved (image). All cx remained negative.
Some dogmalysis offered by this case – – Fluids (probably) helped the pulmonary edema; CPAP/diuresis may have worsened. – IV metop contraindicated in hypotension? Not in this case – Sometimes you may have to diurese someone who recently had DLVOTO, as discussed above.
This case highlights the cognitive flexibility required to deal with hemodynamic puzzles. One thing I would’ve done different is be more aggressive with metop early on as it made a huge difference with DLVOTO. This was quite a ride. Hope you had fun. Feel free to share!
Much kudos to the treating team, I think this was excellently managed. As Amand says, cognitive flexibility ias absolutely key in assessing hemodynamics, particularly in the grey zones when multiple processes occur and co-exist. Managing this type of case using a recipe-based approach and without POCUS could have let to a poor outcome.
Now the POCUS used in this case is on another level. Very impressive and allowing incredible insight and certainly many potentially clinically useful Doppler analysis tips for LVOTO and LAP assessment.
In the end, I think that there were three pathologies, (a) tamponade physiology, (b) dynamic LVOTO, exacerbated by (c) hypovolemia (diarrhea) I might have approached this differently, had I seen a truly plethoric IVC. In such a case, one can easily see how tamponade physiology would contribute to LVOTO in two ways by creating intracardiac hypovolemia, hence worsening LVOTO both by decreasing LV preload and by the compensatory tachycardia. My first approach would probably have been to drain the pericardial effusion, and reassessing the hemodynamics afterwards, but correcting the intravascular deficit was necessary.
The other important thing this case re-emphasize is that tamponade is not a static diagnosis but a physiological spectrum. For the same given effusion (read intrapericardial pressure – IPP), it is the RA pressure that will determine whether overt tamponade develops. In this patient, it is very likely that a day earlier, there was no frank tamponade, but that after some diarrheal volume loss, the RAP dropped, and now IPP > RAP. It is important to know this because if you have an effusion and a fairly full IVC, one needs to be very careful with anything that can drop the RAP, meaning diuretics and vasodilators, because these can easily turn pre-tamponade into overt shock. And, as this case illustrates so well, you might even end up with LVOTO and pulmonary edema! Which is one of the myriad reasons one should have a basic POCUS exam in every acutely ill patient. These are things a resucitationist needs to know and prepare for.
cheers and thanks again to Dr. Thind!
Philippe






 next april!
next april! next april!
next april!



