
So I have a huge issue the IVC and its ultrasound assessment. For the most part, neither the yay-sayers or the nay-sayers are applying much sound physical principles, as far as I’m concerned.
To assess a patient’s volume status, it may be practical to begin with the sub-xiphoid view of the IVC, since the decision to give fluids or not – especially in emergency situations – can then be taken within the first few seconds of examining the patient. The physiological rationale behind assessing the IVC as a marker for volume responsiveness is simple and solid. As the venous compartment fills, the size of the IVC will gradually increase until it reaches a maximal size of about 20-25mm or even 30mm, depending on physical size and chronicity.
Concomitantly, the phasic respiratory variation will decrease as the venous pressure increases and the effect of varying intrathoracic pressure is no longer felt. At this point, the flat part of the Starling curve of the right ventricle is approaching, and there is little response to volume, and little physiological rationale to support giving more.
Currently, many use a variation of about 20% or more to suggest a volume responsive state in ventilated patients or an inspiratory collapse “sniff test” of 50% or more in spontaneously breathing patients.
There remains controversy around using IVC assessment for volume responsiveness, and with good reason! There are a few important reasons why:
a. technique – First of all, there is the manner in which IVC measurement has been taught: the M-mode measurement of the antero-posterior (AP) diameter during breathing (usually of ventilated patients) about 3 cm below the diaphragm. Although highly practical and reproducible, it has many shortcomings. If we look at the physiology, what changes with cycles of respiration is the volume of blood entering the chest/right atrium. Hence the key variable we are trying to assess is the transient variation in IVC size – which is a volume, not a linear dimension (I’m getting painful flashbacks of using pressure to determine volume!)Hence the use of a single linear measure on one point along the length of the IVC to assess this is inherently flawed. For instance, the figure to the left shows how, if this IVC were to be measured in its AP diameter, the variability may not be that great. In the short axis, however, one can clearly see the significant change in surface area, and hence volume between phases of mechanical ventilation. Also, the IVC is rarely perfectly circular, but often ovoid, and occasionally with the greatest diameter in an anteroposterior axis, making that single AP measurement even less relevant.
b. intrathoracic pressure – Secondly, the variation in intrathoracic or intrapleural pressure (Pip) must be measured, as, for instance, a young and fit patient can generate large changes, which would result in more significant IVC variation, as compared to a frail elderly patient, even if they are on the same point of their Starling curve, invalidating the IVC measurement. All “sniffs” are not created equal.
c. intra-abdominal pressure – Finally, the intra-abdominal pressure (IAP) must also be assessed, since an elevated pressure would decrease the size of the IVC and make that measure no less accurate, but less relevant in terms of representing venous filling.
So what should we do?
Instead, a more global assessment of the IVC volume, measuring short axis area measurements and variation at several points along the IVC would give a much more accurate estimate of IVC volume variation. This is currently being studied by our group.
“Eyeballing the IVC” Attempting to link evidence and physiology, some bedside sonographers’ approach is to take a global look at the IVC in both long and short axis during respiration, while clinically assessing the respiratory effort and the abdominal pressure. This approach is analogous to the “eyeballing” of LV function – versus more formal measurements such as Simpson’s disk method, etc – which has been proven just as accurate with sufficient clinical experience.
The figure below shows an IVC that is about 10-12mm along most of its intrahepatic segment on expiration, and collapses almost completely on inspiration. If this belongs to a patient breathing with little effort and with a soft abdomen to palpation, it is physiologically quite clear that this patient would be fluid responsive. It also shows how impressive the collapse is in the short axis.

For instance, let’s say Patient A is in respiratory distress and using accessory muscles and presents to the ER with a respiratory rate of 35 and a systolic BP of 80. His IVC measures approximately 21 mm in diameter at several points along its axis, and has a brief collapse to about 10 mm with strong inspiratory efforts. His abdomen is soft during inspiration but firm during a prolonged expiratory phase.
Patient B is brought to the ER somnolent with a respiratory rate of 10 and a systolic BP of 80. His IVC measures 15 mm with minimal respiratory variation. His respirations are shallow and his abdomen soft. These two patients show how the IVC assessment needs to be taken in clinical context.
Patient A has some 50% inspiratory collapse of a large IVC in the context of large variations in Pip, whereas Patient B has little variation of a mid-sized IVC in the context of very small variations in Pip. In all likelihood, Patient A is not very volume responsive, while Patient B probably is.
Volume Responsiveness vs. Volume Tolerance – it is critically important to distinguish the difference between these two concepts as they are often misused interchangeably: Volume responsiveness refers to an increase in cardiac output (CO) to a fluid challenge. This is a purely hemodynamic concept. Volume Tolerance refers to whether or not a patient can tolerate volume without clinically significant side effects. This is a complex clinical assessment that should include: -the patient’s plasma oncotic pressure (serum albumin) and level of capillary leak, if present, -the patient’s pathology – is there risk of capillary leak in critical tissues such as lung, brain, abdomen? -the type of fluid being considered (isotonic crystalloid vs hypertonics or colloids/blood products). It is important to distinguish that not all patients who are volume responsive are necessarily volume tolerant.
Volume assessment summary – this issue remains a difficult one, even with the use of bedside ultrasound, because the optimal point for any one patient to be on his or her Starling curve at any one time in different clinical conditions remains elusive. Despite considerable study and several proposed management algorithms, there is no means by which to determine exactly how much of any given fluid is enough, without being too much.
In my opinion… – no direct evidence – keeping an IVC below 20mm and probably below 15mm with significant respiratory variation, if hemodynamics allow, is probably ideal. However, bedside ultrasound allows to clearly identify the cases where fluid is clearly needed and those where fluid is unlikely to benefit. Both of these scenarios are easily and routinely missed by traditional examination. Additionally, when the IVC assessment is done in conjunction with lung ultrasound, it becomes possible to detect early development of pulmonary edema and halt aggressive fluid resuscitation (FALLS Protocol, Daniel Lichtenstein).
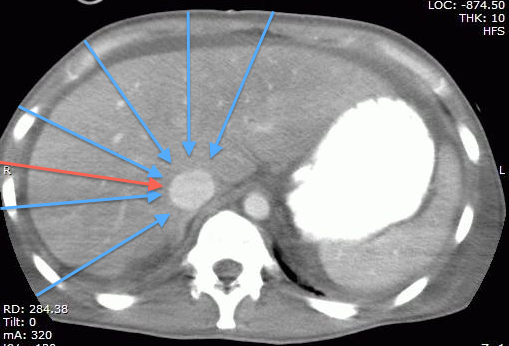
Technical Pearl: the part of the IVC we generally assess being the intrahepatic segment, it is possible to find it almost by scanning through any part of the liver, which happens to provide a great acoustic window. This may be particularly useful when the epigastric area is difficult to access (incision/bandage, drains, in the OR, etc…) or when there is bowel gas in the epigastrium. The figures below show the same IVC, first in a “traditional” epigastric view, then in a view approximately along the plane of the red arrow on the CT scan.

Bottom line?
When assessing volume status, it is absolutely essential to keep the clinical question in mind. It is a great minority of patients who are volume responsive who actually need volume. Normal, healthy humans are very much fluid responsive and fluid tolerant but certainly not in need of any. Much of the current studies and literature focus on assessing volume responsiveness in the setting of shock, which, although arguably the most important, is not the only type of information that can be obtained from the IVC. For instance, as will be discussed in the chapter on congestive heart failure, knowing that your patient is very “full” should prompt further diuresis. If you are dealing with managing severe anasarca, knowing that a patient’s intravascular volume is low may prompt the use of albumin or hypertonics prior to further diuresis to help resorb some of the interstitial fluid. Another critical question to which there is currently no answer is just how much fluid to give to patients in shock. There are many opinions but no certainty. It is common practice to fill a patient in shock until they are no longer fluid responsive, in an effort to avoid or minimize the use or dose of vasopressor medications. There is no study to date that compares a “moderate fluid/early vasopressors” vs “aggressive fluid/avoid-vasopressors-if-possible” approach.
Hopefully this will be answered soon. In light of the clear evidence linking positive fluid balance and mortality, it would seem wise to fill to a “moderate fullness” where some respiratory variation remains, rather than to the point of no longer being fluid responsive. After all, physiologically, the only time humans are really full (>20mm IVC with little or no variation) is in pathological states of congestive heart failure or obstructive shock. So again, when assessing an IVC, keep in mind what your clinical question is and interpret the sonographic data accordingly.
cheers!
Comments:
Marco says:
Thanks Philippe, very interesting post.
Using your great categorization, I think that the worst scenario is represented by a patient who is volume responsive but poorly volume tolerant. In that case, it is important to have clear in mind which is our target: cerebral perfusion, oxygenation and lung extravascular fluid, ventilatory weaning, renal function, avoiding vasopressors or mechanical ventilation, etc…
Very often, maintaining a brain dead heart beating donor, every organ would require a different volemic status, and if all of them are suitable for transplantation you have to compromise.
Just a last thought: as well as “all sniffs are not created equal”, also tidal volumes are very variable. Assessing respiratory variations during protective (low TV) ventilation requires a thoughtful interpretation of the results.
Absolutely. Interpretation of the findings in each individual case is key.
Philippe


